Skeletal muscle undergoes many changes in response to disease and trauma. With the electron microscope, the abnormalities seen at the light level can be characterized and accurately localized, and the variety of changes affecting each organelle identified. The interpretation of the pathological abnormalities observed in a muscle biopsy must take into account several factors, in particular the small sample size, possible artefacts induced by preparation and the non-specificity of the changes.
Only very small areas of tissue can be examined in the electron microscope, and samples may not be representative of the muscle or of the fibre as a whole. Only part of a fibre may be pathologically affected, and frequently isolated and peculiar structures of unknown significance may be observed. Correct handling of samples is also of paramount importance to prevent artefacts. For example, fibres at the edge of blocks may be damaged and show contraction bands, or organelles may swell if the osmolarity and concentration of the buffer are not correct, or the sample is kept in contact with too much saline before fixing. Ideally, muscle should be fixed at resting length for electron microscopy. This is possible for open biopsies by clamping the sample before excision or suturing each end of the muscle strip and securing this to a wooden tongue depressor or other rigid object before fixation. For needle biopsies this is not practicable, but leaving the muscle to rest for 10–15 minutes reduces some of the contraction that occurs with fixation and does not influence the interpretation of the biopsy. Needle biopsies are therefore quite adequate for electron microscopy, particularly for qualitative pathological studies. In general, more ultrastructural information is obtained from studies of longitudinally orientated fibres, particularly with regard to sarcomeric structure. Transverse sections contain more fibres per area, however, and a good impression of fibre size and shape can be obtained from semi-thin 1 μm sections, which in general provide useful information (see Figs. 1.11 and 1.12; ).
The non-specificity of most ultrastructural changes makes it difficult to make a definitive diagnosis by electron microscopy alone. Muscle reacts to disease in a limited number of ways and most abnormalities can occur in a variety of disorders. Nevertheless, consistent patterns can be recognized that assist in making a diagnosis. Accurate diagnosis, however, is possible only when all clinical, histochemical, immunohistochemical, electron microscopic, biochemical and electrophysiological data are considered.
With the developments of immunohistochemistry and molecular analysis the diagnostic role of electron microscopy has diminished, but its main contributions are:
- •
to help decide if a sample is normal or abnormal by looking for any subtle changes;
- •
to clarify the nature of a feature observed at the light level, such as nemaline rods, abnormal mitochondria, core-like areas, or various types of inclusions or accumulated material;
- •
to identify structures only visible at the ultrastructural level, such as nuclear inclusions, the filamentous inclusions of inclusion body myositis or tubuloreticular inclusions in capillaries.
This chapter is intended to illustrate the variety of ultrastructural changes encountered in human muscle biopsies and to serve as a practical guide to the interpretation of the changes. Table 5.1 summarizes many of the abnormalities that occur in human neuromuscular disorders and deals with the individual components of the fibres rather than the changes that occur in specific disorders. Reference to particular diseases is kept to a minimum in this chapter, but in subsequent chapters electron microscopic findings relevant to specific diseases are discussed. We have retained citations of several of the early publications as a reminder of the contribution and pioneering work of morphologists to our understanding of the pathological changes in muscle, before any molecular defects were known. Further details of the occurrence of particular features can be found in reviews by and , which have stood the test of time, and more recent texts such as and .
| 1Sarcolemma |
| Folding Redundant basal lamina Thickening of basal lamina Loss of plasma membrane Abnormal caveolae |
| 2Myofibrils and Cytoskeleton |
| Loss and splitting Hypercontraction I band loss A band loss Ring fibres Cores Filamentous bodies Concentric laminated whorls Z line alterations:
|
| 3Nucleus |
| Central location Changes in shape Changes in chromatin distribution Inclusions Abnormal membrane associated with nuclei |
| 4Mitochondria |
| Abnormal distribution Aggregates Abnormal structure Inclusions |
| 5Membrane Systems |
| Swollen sarcoplasmic reticulum Replication of triads Honeycomb structures Tubular aggregates |
| 6Deposits and Particles |
| Excess glycogen Excess lipid Polyglucosan material Lipofuscin Lipopigment Amyloid Virus-like particles Crystalline material |
| 7Other Unusual Structures |
| Actin accumulation Zebra bodies Fingerprint bodies Curvilinear bodies Reducing bodies Hexagonal crystal arrays Autophagic vacuoles Membranous/myelin-like whorls Dense tubules Mallory body-like inclusions |
Sarcolemma
A common feature is irregularity of the surface of the fibre and folding of the sarcolemma ( Fig. 5.1 ). Such changes are often seen in situations where atrophy occurs. The basal lamina may split away from the plasma membrane and form extensive folds in the extracellular space ( Fig. 5.2 ), or it may appear to be replicated in some areas ( Fig. 5.3 ). Sometimes glycogen is seen between the plasma membrane and basal lamina, and, although this is usually only in small amounts, it can be excessive in glycogen storage diseases ( Fig. 5.4 ).




Redundant basal lamina is considered to be a characteristic of atrophic fibres. This is in contrast to hypotrophic fibres, which have a closely adherent basal lamina. Hypotrophic fibres are believed to be small fibres in which normal growth and maturation have been arrested ( ). It is a term applied to the small type 1 fibres of congenital myopathies such as myotubular (centronuclear) myopathy, but our studies of fibres in this disorder indicate that redundant basal lamina can occur around some small fibres (see Ch. 15 ).
Thickening of the basal lamina also occurs and is found in a variety of neuromuscular disorders ( Fig. 5.5 ). Thickening and duplication of capillary basal lamina are also seen in diseased muscle. It is a feature of diabetes ( Figs. 5.6 and 5.7 ), and thickened basal lamina is a feature of capillaries known as pipestem capillaries, which have been observed in various myopathic conditions including some cases of necrotizing myopathy ( ).



In the recessive Ullrich form of congenital muscular dystrophy caused by defects in collagen VI genes, fibrillar masses near the reticular layer may be seen that probably relate to abnormally assembled collagen VI ( ).
The plasma membrane may also show defects. Focal breaks may be apparent ( Fig. 5.8 ), or in necrotic fibres the plasma membrane may be lost completely ( Fig. 5.9 ). These fibres are then only surrounded by the highly resistant basal lamina. In the search for the underlying cause of Duchenne dystrophy, now known to be dystrophin, considerable emphasis was placed on the observation of damaged plasma membranes ( ), but it is a non-specific finding associated with a particular type of fibre damage and can be found in a variety of disorders ( ).


The plasma membrane also shows changes in disorders affecting proteins of caveolae, in particular caveolin-3 and cavin-1 (also known as polymerase 1 and transcript release factor, PTRF) ( ). With transmission electron microscopy, caveolae appear as small subsarcolemmal vesicles (see Fig. 3.17a ), but when the gene for caveolin-3 is mutated there is impairment of caveolae formation, discontinuity of the plasma membrane, subsarcolemmal vacuoles, papillary projections and disorganization of the T-system openings on the plasma membrane. Cases with mutations in the cavin-1 gene show a marked reduction of caveolae. Caveolin-3 and dysferlin interact, and similar changes in the sarcolemma, including thickening of the basal lamina, may be seen when dysferlin is deficient ( ).
Myofibrils and Associated Cytoskeleton
Loss and alterations in the myofilaments are the most common abnormalities observed in diseased muscle. Their occurrence is widespread, and they have been observed in all classes of genetic and acquired neuromuscular disorders. The degree of myofilament loss and disruption is variable, depending on the nature of the disorder and the area examined. It is one of the most difficult changes to assess because, even in normal muscle, departures from the classical appearance frequently occur. This is shown in Figs. 5.10–5.12 , which are all taken from healthy volunteers and illustrate the variety of features within normal individuals. Myofilament loss in diseased muscle may affect part or the whole of the fibre, and the extent of damage varies from fibre to fibre. Therefore, within a biopsy there are a spectrum of changes, with fibres affected to varying degrees. Focal loss of myofilaments within a fibre may occur ( Fig. 5.13 ), or it may be widespread and cause narrowing of the myofibrils ( Fig. 5.14 ). Care is needed in interpreting focal loss to ensure that changes are not just due to undulation of the myofibrils and differences in the plane of section through sarcoplasm or myofibrils. Excessive splitting of the bundles is often associated with myofibril loss (see Fig. 5.14b ), and the space between the myofibrils is then occupied by sarcoplasmic components, including glycogen, mitochondria, sarcoplasmic reticulum (SR) and T tubules. In rare cases with mutations in both the MuRF1 and MuRF3 genes, subsarcolemmal collections of randomly orientated sarcomeres have been seen ( Fig. 5.15 ).






In severely necrotic fibres, the characteristic myofilament structure is lost completely and replaced by amorphous granular material ( Fig. 5.16 ). These fibres may contain macrophages (see Fig. 5.16 ) and correspond to the pale disrupted fibres seen with histological and histochemical techniques. This appearance is thought to be the end stage of a series of events leading to necrosis ( ). Earlier stages in necrosis are thought to involve the hypercontraction or overcontraction of myofibrils and probably correspond to the round, intensely stained fibres seen with light microscopy. The areas affected by hypercontraction may be focal ( Fig. 5.17 ) or extensive ( Fig. 5.18 ). Clumps of contracted myofibrils form and are interspersed with areas of overstretched filaments. Other organelles in these fibres also show abnormalities, and the tubular systems are often dilated and the mitochondria degenerate. Hypercontraction of myofibrils can be induced artefactually, particularly at the periphery of biopsies where handling may damage fibres. Most hypercontracted fibres seen in diseased muscle, however, are not thought to be artefactual. They are particularly common in Duchenne muscular dystrophy, but also occur in a variety of other neuromuscular disorders.



Selective loss or abnormalities of particular regions of the sarcomere may also occur. Loss of I bands has been reported in some conditions ( ) and is usually associated with loss of the Z line ( Fig. 5.19 ), although remnants of the Z line may sometimes remain. Loss of I bands but retention of A bands with M lines in unusual peripheral fragments of sarcomeres has been reported in a myopathy associated with MuRF1 and MuRF3 mutations (see Fig. 5.15 ) ( ). As stated previously, caution in interpretation is sometimes required if the plane of section has not passed completely through the whole myofibril. Sections through contracted or disorientated myofibrils may pass through filaments at one point and sarcoplasm at another, thus giving the false impression of loss. Loss of A bands ( Fig. 5.20a ) is a rare finding in diseased human muscle, but a number of cases have been reported and we have observed isolated fibres with this abnormality in several conditions, including systemic lupus erythematosus, congenital muscular dystrophy and carriers of Duchenne muscular dystrophy. It is most commonly found in patients with acute quadriplegic myopathy (‘critical care myopathy’) on high doses of glucocorticoids and neuromuscular blocking agents (see Ch. 23 ) ( ). In some cases the A band loss is only partial and may only affect a few sarcomeres, leaving the Z line intact ( Fig. 5.20b ). Mutations in the gene encoding slow myosin ( MYH7 ) can result in the presence of hyaline bodies . These areas of fine, rather granular material are devoid of organelles but contain slow myosin; they are discussed further in Chapter 15 .


Myofibrils can undergo varying degrees of disorganization, disorientation and disruption. In some fibres, the normal striation pattern is lost completely but sarcomeres are still identifiable ( Fig. 5.21 ). In others, only certain regions of the fibre are disorientated: for example, in ring fibres ( Figs. 5.22 and 5.23 ). These unusual fibres have one or more peripheral myofibrils running at right angles to the normal axis of the fibre. The disorientated zone of myofibrils may be immediately beneath the sarcolemma (see Figs. 5.22 and 5.23 ) or be separated by an area of sarcoplasm that contains very few myofilaments. These areas are known as sarcoplasmic masses ( Fig. 5.24 ). In some ring fibres, the disorientated myofibrils may not be peripheral throughout the fibre and bands of myofibrils at right angles to the remaining myofibrils can be seen to traverse the fibre (see Fig. 5.24 ). Ring fibres are non-specific but are particularly common in myotonic dystrophies.




In cap myopathy focal peripheral areas of disorganized myofibrils are seen (see Ch. 15 ). They have been associated with defects in several genes associated with a congenital myopathy, including ACTA1, TPM2 , TPM3 and TTN . They probably form part of the pathological spectrum associated with nemaline myopathies.
Restriction of myofilament disruption to focal areas is found in the formation of cores, minicores and target fibres. Cores , such as those found in central core disease caused by mutations in the RYR1 gene, can be central or peripheral and run most of the length of the fibre (see Ch. 15 ). They are characterized by a variable degree of myofibril disorganization and Z line streaming and a scarcity or absence of mitochondria. recognized two pathologically distinct types of core in central core disease: the structured and the unstructured core. In the structured core, the striation pattern is preserved but the core area is slightly contracted compared with the surrounding myofibrils and shows a reduction in the number of mitochondria ( Fig. 5.25 ). This type of core retains its myosin adenosine triphosphatase (ATPase) staining in contrast to the unstructured core, in which it is lost or reduced. The banding pattern in unstructured cores is not discernible, and there is usually marked myofibrillar disruption and large amounts of smeared Z line material, but mitochondria are sparse or absent (see target fibres later). Some authors report the occurrence of both types of cores in the same biopsy ( ), but considered this unusual and that it is more common to find only one type of core in a given patient with central core disease. In a case of central core disease referred to us, some cores could not easily be categorized as structured or unstructured and were intermediate between these types ( Fig. 5.26 ). The myofibrils of these cores showed degenerative changes but had recognizable striations. The extensive disorganization found in unstructured cores was absent. Tubular profiles were also clearly visible in these cores. These features of different types of cores probably reflect different stages of myofibrillar disruption within a spectrum that can be seen within a single biopsy, and the distinction is not of diagnostic relevance.


Other clinically distinct conditions are characterized by the presence of multiple minicores ( ; see Ch. 15 ). These cores are small, focal areas of disruption and affect only a few sarcomeres ( Fig. 5.27 ), or they may extend over a wider area involving several sarcomeres and myofibrils ( Fig. 5.28 ), but they are not usually as extensive as those in central core disease. Minicores are characterized by irregularities and smearing of the Z line and disorganization of the myofibrils. Central core disease and multi-minicore disease, caused by mutations in the genes for the ryanodine receptor 1 ( RYR1 ) and selenoprotein N1 ( SEPN1 now known as SELENON ), respectively, gained their names from the extent of the pathological abnormality (see Ch. 15 ), but cores of varying dimension occur in association with defects in both genes. They are not specific for these disorders, however, and cores, particularly minicores, can be found in a variety of neuromuscular disorders. Figs. 5.27 and 5.28 were from two members of the same family with a mutation in the MYH7 gene. Electron microscopy is useful for identifying subtle core-like lesions that are not easily visible with light microscopy.

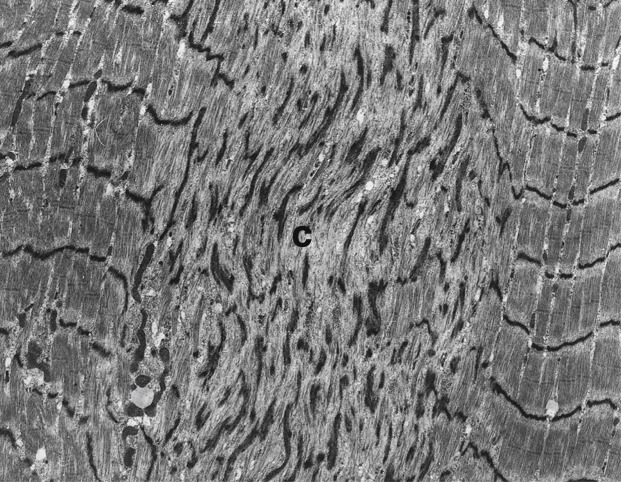
In some cores the myofibrillar disruption may be minimal and only misalignment of the striations is seen in comparison with adjacent myofibrils. These areas have a scarcity of mitochondria ( Fig. 5.29 ) and may correspond to unevenness of oxidative enzyme stain. Caution in identifying such areas is needed, however, in samples not fixed at resting length, as variable contraction of myofibrils may lead to misalignment.

Target fibres ( ) have three concentric zones that can be identified with both the light and the electron microscope and are a feature of denervated muscle: the outer zone is essentially normal except for occasional areas of Z band streaming; the intermediate zone contains myofibrils with mild Z line irregularities and swollen SR; and the central zone is markedly disrupted with extensive Z line material similar to unstructured cores ( Fig. 5.30 ). If the intermediate zone is not clearly defined, the fibres are termed targetoid .

A congenital disorder has been described in which a population of ‘trilaminar fibres’ with zones was found ( ). In these fibres the outer zone contained mitochondria, loosely packed filaments, glycogen, ribosomes and tubular profiles; the intermediate zone consisted of a few organized myofibrils with characteristic striations; and the inner zone contained mitochondria, glycogen, osmiophilic material resembling Z line material and loosely packed filaments.
Other unusual non-specific structures which are thought to be of myofibrillar origin are filamentous bodies and concentric laminated bodies. Filamentous bodies are composed of tightly packed actin-like filaments ( Fig. 5.31 ). Within a biopsy they are infrequent and when present they are often subsarcolemmal but can occur in other regions of the fibre. They have been identified in a variety of neuromuscular disorders and we have also often seen them in carriers of Duchenne dystrophy. They have also been recorded in normal human muscle. They were a particular feature in some rare cases, but it is not clear if they are associated with a disease entity ( ).

Concentric laminated bodies are cylindrical structures of 3–25 concentric laminae, the centre of which may contain glycogen ( Fig. 5.32 ). The laminae are 6–8 nm thick, with a spacing of about 7.5 nm. They are considered by some workers to be of myofibrillar origin ( ), whereas other workers believe they are derived from mitochondria ( ). They are non-specific.

Z Line
Z line streaming is another very common structural alteration associated with myofibrillar damage in diseased muscle ( Fig. 5.33 ). The number of sarcomeres involved is variable, and some Z line streaming can be found in normal muscle. In areas adjacent to capillaries, focal areas of myofibrillar disruption are common. The Z line may also show thickening ( Fig. 5.34 ), irregularities (see Fig. 5.33 ), duplication ( Fig. 5.35 ) or loss (see Fig. 5.33 ). It is also thought that the Z lines give rise to the dense rod-like structures that characterize all forms of nemaline myopathy ( ; Fig. 5.36 ), and some of the electron-dense material in myofibrillar myopathies may be derived from Z line material (see Ch. 16 ).



Related posts:
 The Procedure of Muscle Biopsy
Histological and Histochemical Stains and Reactions
Muscular Dystrophies and Allied Disorders II: Limb-Girdle Muscular Dystrophies
Muscular Dystrophies and Allied Disorders I: Duchenne and Becker Muscular Dystrophy
Ion Channel Disorders
Myofibrillar Myopathies and Other Myopathies with Rimmed Vacuoles
The Procedure of Muscle Biopsy
Histological and Histochemical Stains and Reactions
Muscular Dystrophies and Allied Disorders II: Limb-Girdle Muscular Dystrophies
Muscular Dystrophies and Allied Disorders I: Duchenne and Becker Muscular Dystrophy
Ion Channel Disorders
Myofibrillar Myopathies and Other Myopathies with Rimmed Vacuoles
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


