Secondary Osteoarthritis
H. Ralph Schumacher Jr.
Lan X. Chen
Joseph Buckwalter
In increasing percentages of patients, osteoarthritis (OA) can now be classified as secondary on the basis of an identifiable congenital, developmental, traumatic, or systemic disease that appears to explain the degenerative changes in the articular cartilage. All diseases considered in this chapter have clinical, radiologic, and pathologic features in common with “idiopathic” OA to varying degrees. However, there are also unique features that suggest each underlying cause. These distinguishing features are emphasized, along with brief descriptions of each disease process. Although osteoarthritic changes associated with these underlying diseases are emphasized, other musculoskeletal symptoms produced by these diseases that may be confused with osteoarthritic manifestations are also described. The systemic diseases causing arthritis deserve major emphasis because 1) in some patients, the OA may be an early or, even initial, clue to a potentially dangerous and treatable systemic disease; 2) these secondary osteoarthritides may have specific therapies, in contrast to the largely symptomatic treatment used in most osteoarthritides; and 3) the mechanisms identified in these examples of secondary OA may provide helpful clues to mechanisms in idiopathic disease.
Specific treatments of underlying diseases or diseases related to them are also discussed in this chapter. Unless otherwise noted, symptomatic and general therapy is as described in Section III for general management of OA.
Systemic Metabolic Diseases
Hemochromatosis
Hemochromatosis is a chronic disease characterized by excess iron deposition and fibrosis in a variety of tissues. Most hemochromatosis is idiopathic. Ninety percent of idiopathic cases are associated with homozygosity for a tyrosine substitution at position 282 on the HFE gene.1 There are a variety of other less common mutations.2 This is not a rare disease. Estimated incidences of clinically detectable disease have varied; homozygosity for the gene in white populations is about 1 in 500. Mutations and disease are much less common in Blacks and Asians.3 Frequent manifestations are hepatomegaly and cirrhosis, increased skin pigmentation (in large part because of increased melanin), diabetes, other endocrine deficiency, and cardiomyopathy. Iron overload usually requires many years to develop, so that most patients have onset of symptoms between the ages of 40 and 60 years. Hemochromatosis is uncommon in premenopausal women, presumably because of menstrual blood loss. The largest iron deposits are in the liver, and biopsy is frequently performed for diagnosis. Elevation of fasting serum iron levels with saturation of iron-binding capacity greater than 62% or elevated serum ferritin levels can help suggest the diagnosis. Genetic testing can be supportive. Liver function abnormalities are often minimal, even with advanced hepatic deposition of iron.
OA-like changes in hemochromatosis were first described in 19644 but have since been recognized to occur in 20% to 50% of patients; one study showed radiographic changes in 81%.5 Age at onset of the arthritis has varied from 26 to 70 years but is most common in the fifth decade. Arthritis generally coincides closely with the onset of other manifestations of hemochromatosis but may antedate other findings and be the first clue to the disease.6,7
The hands, knees, and hips are most commonly involved, although virtually any joint, including those in the ankles and feet,8,9 can be affected. Helpful in diagnosis is the characteristic involvement of metacarpophalangeal (MCP) joints as well as proximal interphalangeal (PIP)
and distal interphalangeal (DIP) joints with a firm, bony, and often only mildly tender enlargement that is different from that seen in rheumatoid arthritis (Fig. 13-1). Involvement of the second and third MCP joints is particularly characteristic, although it can also occasionally be seen in patients with generalized OA, in manual laborers, and in association with calcium pyrophosphate deposition disease. Joints are stiff and become limited in motion, but morning stiffness is not prominent.
and distal interphalangeal (DIP) joints with a firm, bony, and often only mildly tender enlargement that is different from that seen in rheumatoid arthritis (Fig. 13-1). Involvement of the second and third MCP joints is particularly characteristic, although it can also occasionally be seen in patients with generalized OA, in manual laborers, and in association with calcium pyrophosphate deposition disease. Joints are stiff and become limited in motion, but morning stiffness is not prominent.
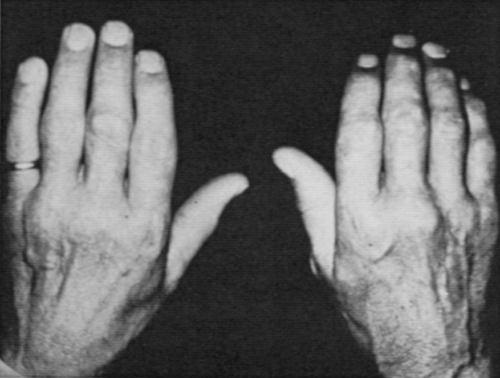 Figure 13-1 Bony osteoarthritis-like enlargement of some of the distal interphalangeal, proximal interphalangeal, and metacarpophalangeal joints in hemochromatosis. |
Joint effusions have been noninflammatory, with leukocyte counts less than 2000/mm2, except during the infrequent attacks of associated pseudogout. Cells are predominantly mononuclear and occasionally contain iron on staining with Prussian blue.10 Measurements of iron levels in synovial fluid are comparable to those in serum.
Synovial tissue shows a striking deposition of iron that is most prominent in the synovial lining cells and, as seen by electron microscopy, is actually greatest in the type B or synthetic cells.11 By light microscopy, the iron is golden and may be missed unless it is specifically looked for. Other synovial changes are only mild lining cell proliferation, fibrosis, and scattered chronic inflammatory cells. Although few cases have been studied, iron is also demonstrable in the chondrocytes of articular cartilage and at the line of ossification. There are degenerative changes in cartilage.12 All cartilages studied to date by electron microscopy have also shown either apatite or calcium pyrophosphate dihydrate (CPPD) crystals, which may be important in pathogenesis.12
Radiographic studies show the characteristic joint distribution that often includes MCP joints. There is joint space narrowing and irregularity; subchondral sclerosis; and often large cystic erosions, hook-like bone proliferation, and even subluxation (Fig. 13-2). In up to 60% of patients, chondrocalcinosis and periarticular soft tissue calcification are seen. Aside from the distribution and frequency of calcifications, the involvement at the hips and other sites seems indistinguishable from idiopathic OA. Three cases have been reported with aseptic necrosis at the hip.13 Whether this underlies some hip OA in these patients needs further study.
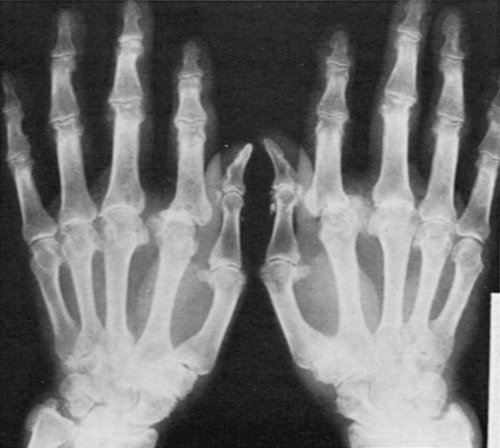 Figure 13-2 Radiograph of the hands in hemochromatosis showing joint space narrowing, subluxations, subchondral cysts, periarticular sclerosis, spurs, and soft tissue calcifications, all most prominent at the second to fourth metacarpophalangeal joints. |
Musculoskeletal manifestations other than OA are also seen. As noted earlier, pseudogout attacks can occur from CPPD crystals. Apatite may also be involved in some crystal-induced arthritis. Osteopenia is seen and may be related to the cirrhosis or to androgen or other endocrine deficiency.
Mechanisms involved in the arthritis are not established, although there are intriguing possibilities.14 Iron deposition in the chondrocytes could alter the proteoglycans, collagen, or enzymes released by these cells, leading to the degenerative change in the matrix. Iron could promote the formation of toxic free radicals or could bind directly to some proteoglycans and alter their function, as it has been shown to do in vitro. In vitro iron can downregulate prostaglandin E2 production by synovial fibroblasts. The balance of this effect on these cells could have deleterious or beneficial results.15 Iron, in vitro, also inhibits the enzyme pyrophosphatase and could thus contribute to the deposition of CPPD crystals. Because the iron and calcium crystal depositions are not spatially related, iron would appear to promote calcification in some indirect way, or alternatively, the calcifications may be a result of unrelated mechanisms or related inherited factors. Although cartilage appears to be primarily involved in the OA, the synovium also has heavy deposits of iron, and stimulation of release of cytokines or enzymes from the synovium might contribute to the process. Synovial siderosis might alter the clearance of factors involved in calcification.16 The possibility has been raised that the HFE mutation may predispose to hand or other OA even without iron overload.17 This has not been confirmed to date for hip and knee OA.18
Hemochromatosis can be treated and many systemic features reversed or prevented by removal of excess iron
with intensive and continued phlebotomies or with the chelating agent deferoxamine. Alcohol, which increases the risk of liver damage, and vitamin C ingestion, which increases iron absorption, should be avoided. Once established, joint disease has not been reversed, and in fact, some patients have had their first joint symptoms or exacerbations of arthritis after phlebotomy. In vitro studies suggest that iron could even inhibit CPPD deposition so that depletion of iron might possibly exacerbate chondrocalcinosis.19 Episodes of acute crystal-induced arthritis should be watched for and can be treated with nonsteroidal anti-inflammatory agents. Prosthetic hip and knee replacements have been successfully performed for chronic changes. Family members of all patients with hemochromatosis should be screened for iron overload as early treatment may prevent arthritis and other manifestations.
with intensive and continued phlebotomies or with the chelating agent deferoxamine. Alcohol, which increases the risk of liver damage, and vitamin C ingestion, which increases iron absorption, should be avoided. Once established, joint disease has not been reversed, and in fact, some patients have had their first joint symptoms or exacerbations of arthritis after phlebotomy. In vitro studies suggest that iron could even inhibit CPPD deposition so that depletion of iron might possibly exacerbate chondrocalcinosis.19 Episodes of acute crystal-induced arthritis should be watched for and can be treated with nonsteroidal anti-inflammatory agents. Prosthetic hip and knee replacements have been successfully performed for chronic changes. Family members of all patients with hemochromatosis should be screened for iron overload as early treatment may prevent arthritis and other manifestations.
Wilson Disease (Hepatolenticular Degeneration)
Wilson disease is an uncommon familial disease associated with a variety of mutations in a gene (ATPTB) on chromosome 13 q14.3.20 The most frequent mutation is a substitution of glutamine for histidine at amino acid 1069.20 The disease is characterized by the Kayser-Fleischer ring, consisting of brown pigment at the corneal margin; cirrhosis; and basal ganglion degeneration leading to tremor, rigidity, or other neurologic problems. Many patients also develop renal tubular acidosis. The onset of symptoms occurs between the ages of 4 and 50 years. A disorder of copper metabolism can be demonstrated by an increase in urinary excretion of copper and a general decrease in the serum copper-binding protein ceruloplasmin. Copper concentration is increased in liver, brain, and other tissues. A rapid polymerase chain reaction (PCR) test can be used to document the most common mutation in ATPTB.20
Arthropathy is rare in children but occurs in up to 50% of adults.21,22 The OA may be asymptomatic despite radiographic findings or may be markedly symptomatic with worsening on activity. More commonly involved joints have been the wrists, elbows, shoulders, hips, and knees and, occasionally, the fingers. The early age at onset and the prominent involvement of the wrists in many patients suggest a difference from primary OA.14,21,22
Joint effusions are usually small and consist of clear, viscous fluid. Leukocyte counts have been approximately 200 to 300/mm3 with predominantly mononuclear cells. Synovial biopsy specimens have shown mild lining cell hyperplasia and few chronic inflammatory cells.14,22,23 Cartilage has been examined in four patients and was shown to contain copper by energy-dispersive elemental analysis in two.23 Copper has also been found in synovium, where it might alter cytokine and protease production.24,25 CPPD crystals were not noted in our patients but have been reported in an intervertebral disk.26 Joint hypermobility occurred in 9 of 32 patients in one series.27
Radiographic joint findings have included subchondral bone fragmentation and sclerosis, subchondral cysts, cortical irregularity, cartilage space narrowing, periarticular cysts, vertebral wedging,14,21,22,25 osteochondritis dissecans,27 and severe chondromalacia patellae.22 Periarticular calcifications are common. Some calcifications have been thought to represent bone fragments, and chondrocalcinosis has been described.23
No correlation has been found between total disease severity, spasticity or tremor, osteopenia, or liver or renal disease and the arthritis. Experimental studies of only short-term copper loading have not produced an arthropathy. Although the nature of most joint calcifications is not yet known, McCarty and Pepe28 showed that cupric (as well as ferrous) ions could inhibit pyrophosphatase in vitro, suggesting a possible cause for deposition of CPPD crystals. In vitro copper loading of articular chondrocytes has altered matrix synthesis and caused increased collagen production.29
Wilson disease is associated with osteopenia in 25% to 50% of patients in different series. It is usually asymptomatic but can be painful in the presence of pathologic fractures. Some of the bone demineralization results from definite rickets or osteomalacia attributed to the renal tubular disease.
This is a treatable disease; penicillamine appears to be the most effective chelating agent for mobilizing copper from the tissues, although zinc may also be used. Treatment is continued for life. Although neurologic improvement is often reported, there is no evidence that the established arthropathy has been helped; in fact, some penicillamine-treated patients have subsequently developed the OA. Whether early diagnosis and treatment can prevent the arthritis is not yet known. Family members should always be checked to try to establish an early diagnosis. Penicillamine seems occasionally to produce polymyositis, lupus, inflammatory polyarthritis,27 and other immunologic syndromes.
Ochronosis
Ochronosis is the result of a hereditary deficiency of a liver enzyme, homogentisic acid oxidase; the defect is now mapped to chromosome 3q with a wide variety of mutations reported.14,30,31,32 Lack of this enzyme allows accumulation of homogentisic acid, which, when excreted in large amounts, imparts a dark brown or black color to the urine. This is termed alkaptonuria. Freshly passed urine usually appears normal but darkens with standing or with alkalinization. This inherited defect is thought to be transmitted as a simple autosomal recessive.
Ochronosis occurs when polymers of homogentisic acid become deposited in connective tissue. The exact mechanisms of affinity of homogentisic acid for connective tissue are not known. Deposition is reversible until the homogentisic acid is polymerized. Mechanisms of tissue injury may include a demonstrated inhibitory effect of homogentisic acid on in vitro chondrocyte growth.33 Ochronotic pigment is black when it is viewed grossly in masses in tissues; when it is seen in thin histologic sections under the light microscope, it is ocher or golden yellow.
Infants may have black staining of diapers. By the fourth decade, ochronotic pigment becomes detectable as a blue-black hue in the external ear cartilage or tympanic
membrane, as scleral pigmentation, or as malar and other cutaneous darkening. Pigment deposits in the mitral and aortic valves can deform the leaflets and cusps, producing murmurs in 15% to 20% of patients. Calcified prostatic calculi containing ochronotic pigment occur in a large percentage of men with ochronosis.
membrane, as scleral pigmentation, or as malar and other cutaneous darkening. Pigment deposits in the mitral and aortic valves can deform the leaflets and cusps, producing murmurs in 15% to 20% of patients. Calcified prostatic calculi containing ochronotic pigment occur in a large percentage of men with ochronosis.
Deposition of the pigment in intervertebral disks and in articular cartilages leads to degenerative disk disease and peripheral arthropathy. The majority of patients older than 30 years develop spondylosis, which may present with low back stiffness and aching or, in about 15% of patients, herniation of a lumbar nucleus pulposus. Involvement of the dorsal and cervical spine occurs only later. Symptoms may be minimal despite prominent radiographic changes.
Peripheral arthropathy generally occurs later and is milder than the spondylosis. The knees, shoulders, and hips are most commonly involved.35 In peripheral joints, symptoms may antedate detected radiographic changes. As with other forms of OA, symptoms are predominantly pain, crepitation, limited motion, and stiffness.
Joint effusions occur in about 50% of involved knees. Synovial fluid is clear, viscous, and yellow. On occasion, dark specks of ochronotic cartilage can be seen floating in the fluid.36,37 Cartilage fragments seen microscopically in joint fluid are golden yellow. Leukocyte counts are generally in the noninflammatory range, with counts from 112 to 700/mm3; mononuclear cells predominate. Joint fluids have been described with CPPD crystals without inflammatory reaction or with acute attacks of pseudogout superimposed on the degenerative arthropathy.38 Although small amounts of homogentisic acid occur in joint fluid, the amount is insufficient to cause darkening with alkalinization.35
The earliest radiographic changes suggestive of ochronosis are calcification and even ossification in the lumbar intervertebral disks. Hydroxyapatite has been identified as the calcium salt in the disks. Although typical of ochronosis, disk calcification (Fig. 13-3) is not diagnostic because it has also been seen with CPPD deposition disease, hemochromatosis, chronic respiratory paralytic poliomyelitis, ankylosing spondylitis, acromegaly, amyloidosis, tuberculosis, and trauma or without any identifiable systemic disease. The calcification in ochronosis is followed later by disk space narrowing; osteophytes tend to be small but may bridge occasional vertebrae. The sacroiliac joints can show narrowing but do not fuse; typical syndesmophytes like those found in ankylosing spondylitis are not seen.
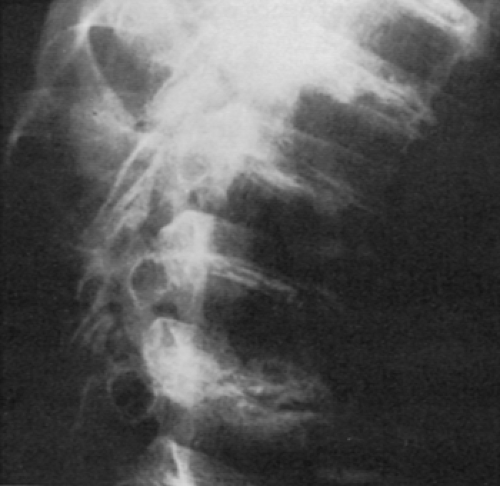 Figure 13-3 Intervertebral disk ossification in ochronosis. |
On radiographic examination, peripheral joints do not differ in appearance from those in other forms of OA except in the distribution of ochronosis. Ochronosis tends to involve the larger joints and spare or only mildly involve the hands and feet. Protrusio acetabuli has been reported in one case.35 Loose bodies occur in some peripheral joints.35 Chondrocalcinosis may be seen. As in the spine, the osteophytes tend to be small.
Ochronotic pigment deposition is initially in the deeper and midzone cartilage but eventually produces grossly visible diffuse blackening of cartilage (Fig. 13-4). This cartilage is so friable that it progressively erodes, with fragments breaking loose into synovial fluid. Pigmented shards become embedded in synovial membrane (Fig. 13-5). Cartilage collagen and associated substances appear to be primary sites of the pigment deposition.35,39 Ochronotic pigment is probably secondarily phagocytosed by chondrocytes and synovial cells. Because chondrocytes show some degenerative changes in virtually all cases studied by electron microscopy, they may also be affected in some way early in the disease.35 The matrix ochronotic pigment and the cellular changes result in a dramatically friable cartilage that predictably degenerates in early middle age. Joint loose bodies appear to arise from osteochondrometaplasia around the ochronotic shards embedded in synovium.35
 Figure 13-4 Blackening of the knee meniscus in ochronosis. |
No satisfactory therapy for the enzymatic defect has yet been developed. Unfortunately, a diet low in phenylalanine and tyrosine precursors has been too unpalatable to demonstrate whether it might have any long-term clinical benefit, although urinary homogentisic acid levels can be
decreased with it.14,35 High doses of ascorbic acid, although they do not decrease total urinary homogentisic acid, have been reported to inhibit binding to connective tissue in experimental alkaptonuria of rats.40 High levels of ascorbic acid in vitro can prevent the inhibitory effect of homogentisic acid on chondrocyte growth.33 Nitisinone is an experimental agent that has had initial clinical studies and has been shown to decrease homogentisic acid levels.40a Corrective orthopedic measures have been effective and without any special problems.41,42
decreased with it.14,35 High doses of ascorbic acid, although they do not decrease total urinary homogentisic acid, have been reported to inhibit binding to connective tissue in experimental alkaptonuria of rats.40 High levels of ascorbic acid in vitro can prevent the inhibitory effect of homogentisic acid on chondrocyte growth.33 Nitisinone is an experimental agent that has had initial clinical studies and has been shown to decrease homogentisic acid levels.40a Corrective orthopedic measures have been effective and without any special problems.41,42
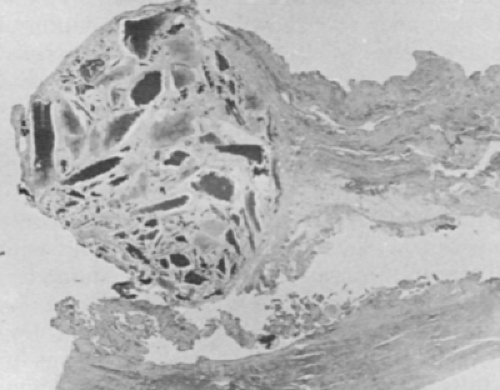 Figure 13-5 Golden brown pigmented shards from the friable cartilage embedded in synovium in ochronosis. |
Gaucher Disease
Gaucher disease is an inherited metabolic disease characterized by the accumulation of glucocerebroside in distinctive Gaucher cells, most prominently in the liver, spleen, and bone. The glucocerebroside deposits occur because of a deficiency of the enzyme glucocerebrosidase.43 The disease is more common in, but not restricted to, people of Ashkenazi Jewish background. Clinical severity can vary widely. Different clinical types with different prognoses have been defined. Some individuals are disabled by the age of 30 years, whereas others lead relatively symptom-free lives to old age.44,45 Common findings in adults are splenomegaly, hepatomegaly, anemia and thrombocytopenia (caused by hypersplenism and, occasionally, marrow replacement), pingueculae, and bone marrow expansion causing such findings as the Erlenmeyer flask appearance of the distal femur. Neurologic problems are more common in children. The age at onset and severity tends to correlate with the degree of glucocerebrosidase deficiency.43
Definitive diagnosis can be made by bone marrow biopsy or by biochemical study of leukocytes detecting low levels of beta glucosidase. Gaucher cells are large reticuloendothelial cells with profuse, “wrinkled,” pale pink cytoplasm on hematoxylin-eosin staining and with one or more small nuclei. Electron microscopy shows that the cytoplasm of these cells is occupied by membrane-bound inclusions filled with tubular structures typical of glucocerebroside. Acid phosphatase is demonstrable within the tubules in some vacuoles. Chemical analysis also shows iron and other components in the vacuoles.43 Gaucher-like cells are not diagnostic of Gaucher disease; they have also been seen in thalassemia46 and chronic myelogenous leukemia.47
Elevated serum levels of acid phosphatase are observed and may be helpful in suggesting a diagnosis of Gaucher disease. Increased angiotensin-converting enzyme48 and relative factor IX deficiency have been reported.
The degenerative arthritis in Gaucher disease follows marrow infiltration, aseptic necrosis, or pathologic fracture,49 with resulting joint distortion. It is most common in the hip but has also been seen in the shoulders and knees. Joint space narrowing is secondary.
Synovial fluid has rarely been examined. In one patient with pathologic fractures of tibial plateaus, joint fluid was clear yellow and contained 600 white blood cells, 7050 red blood cells, and no crystals under polarized light.49 Actual infiltration of Gaucher cells into cartilage has not been noted.
In addition to aseptic necrosis, radiographic changes of the skeleton include demineralization and cortical thinning as a result of the medullary expansion, foci of sclerosis, and pathologic fractures. Epiphyseal and diaphyseal areas of long bones are prominently involved. Shafts of long bones may be widened with the infiltrative process, as is most typically described in the distal femur.
Not all musculoskeletal symptoms are caused by the OA; they also appear to result from pathologic fractures, episodic periostitis or painful bone crises possibly due to ischemia (often with fever) that may be difficult to distinguish from osteomyelitis50 and deep aching bone pain.51 Many bone lesions detected radiographically are asymptomatic. Amyloidosis52 has been noted in two reports in association with Gaucher disease and might offer a secondary cause for musculoskeletal problems.
Reconstructive joint surgery,50 including total hip replacement, has been successful, but hemorrhage has been an important complication. Increased postoperative infections have been reported.
Replacement of the deficient enzyme has been performed, but how effective it will be for bone disease is not known.53 Other theoretical treatments such as gene therapy, extracorporeal degradation of the glucocerebroside, stimulation of residual endogenous enzyme, and alteration of other normal related enzymes have been considered and may be clinically possible in the future.43,54
Hemoglobinopathies
Sickle Cell Disease
Sickle cell disease is by far the most common hemoglobinopathy associated with musculoskeletal manifestations.55 Homozygous sickle cell disease is an inherited disease most common in Blacks and caused by a substitution of valine for glutamic acid as the sixth amino acid in the β-chain of hemoglobin. This results in sickling of erythrocytes, which presumably occludes small vessels, causing painful crises and bone lesions.56 Other manifestations
described include hemolytic anemia, renal involvement with hyposthenuria, leg ulcers, hyporegenerative crises, increased infections (especially with salmonellae), and a variety of rheumatic or bone and joint problems.
described include hemolytic anemia, renal involvement with hyposthenuria, leg ulcers, hyporegenerative crises, increased infections (especially with salmonellae), and a variety of rheumatic or bone and joint problems.
Diagnosis is generally made by hemoglobin electrophoresis or with allele specific oligonucleotide probes. Hemoglobin S comprises 76% to 100% of hemoglobin in homozygous sickle cell disease.
Aseptic necrosis appears to be the basis for any OA in these patients.55 As with other causes of aseptic necrosis, homozygous sickle cell disease, sickle cell-hemoglobin C disease, sickle cell-thalassemia, and possibly sickle cell trait57,58 can lead to OA as a result of the incongruity of the joint space and loss of normal bony support for the articular cartilage. This is most common at the hip but can also involve the spine, knee,59 shoulder60 (Fig. 13-6), and occasionally other joints.
In addition to aseptic necrosis, radiographic changes in sickle cell disease can include coarse trabeculae with hair-on-end appearance in the skull, osteopenia, vertebral indentations, medullary infarctions, and periosteal elevation.
Other bone and joint problems in sickle cell disease that are clearly more typical than OA for this disease include infarction of bone away from joints, hyperuricemia and occasional gout,58 the “hand-foot syndrome” in young children, osteomyelitis and rare septic arthritis, muscle necrosis, and acute joint effusions, usually but not always with low leukocyte counts.55,61 An element of synovitis for which there is no clear explanation62 and a diffuse chondrolysis 63 can damage cartilage and contribute to the later development of OA.
Total hip and knee arthroplasties55,64,65 have been performed for the secondary OA after avascular necrosis and have been highly successful. Preoperative transfusions have been used to decrease the chances of sickling during surgery. Tourniquets should be avoided to reduce anoxia and stasis, which contribute to thrombosis. Good hydration and efforts during anesthesia to avoid any hypoxia and acidosis are also warranted.64
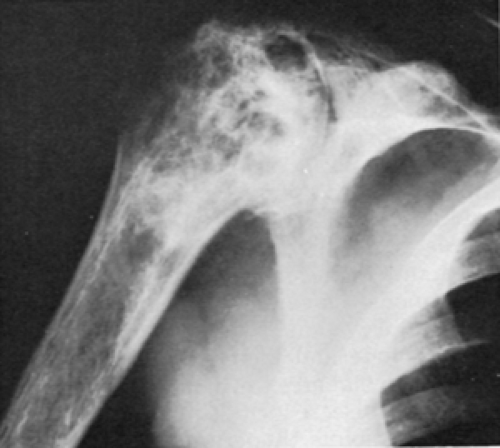 Figure 13-6 Severe marrow infarctions at the shoulder with associated OA in sickle cell disease. |
Thalassemia
β-Thalassemia describes a group of inherited disorders of hemoglobin synthesis resulting in a relative decrease in β-chains. Hemoglobin α-chains accumulate, producing unstable hemoglobin, Heinz bodies, and hypochromic microcytic erythrocytes. Early erythrocyte death results in marrow expansion and splenomegaly. Levels of hemoglobin F or A2 are elevated.
Frequent transfusions are required and often lead to secondary iron overload. Thalassemia major with severe anemia often leads to death in the second or third decade. Milder thalassemia minor may be asymptomatic and may not require therapy.
OA has been described as developing prematurely in thalassemia major and minor. Weight-bearing joints (including the ankles) were predominantly involved in one series, but shoulders, wrists, and elbows have been equally affected in other studies.66,67,68,69,70 The speculation is that marrow hyperplasia may weaken the subchondral bone and allow microfractures that then alter the normal support required by the articular cartilage.69,70 Osteomalacia has been confirmed in the areas of microfractures.70 Multiple transfusions and iron overload might contribute to osteoarthropathy in some patients, as is described in hemochromatosis,71 but early OA has also been described without iron overload.69 Juxta-articular osteopenia and bone cysts67 may be seen on radiographs. Osteonecrosis has been reported, but whether it is increased in incidence has not been established.68,72 Widened medullary spaces with thin cortices, coarse trabeculations, and microfractures are seen in bone with marrow expansion. Synovial fluid, when studied, has been noninflammatory. Tophaceous gout has been described.73
Dull, aching pain, especially at the ankles, has been described after strenuous exercise.70 Because the cartilage space was normal on radiography, the pain was attributed to the periarticular bone involvement.70
No specific treatment is available for the established OA caused by thalassemia. Transfusions may decrease excessive erythroporesis with its associated marrow expansion and bone loss. Bone marrow or stem cell transplantation has been used for some patients for the thalassemia.74 Total hip replacements have been performed.75
Ehlers-Danlos Syndrome and Other Joint Hypermobility
The Ehlers-Danlos syndrome consists of a group of heritable disorders of connective tissue76 with features that include hypermobility of joints (Fig. 13-7), hyperextensible skin, poor wound healing, bruising, and cigarette paper scars. At least seven different types of Ehlers-Danlos syndrome have been identified, with clinical differences, different inheritance, and, in some cases, identified biochemical defects.
The most serious type of Ehlers-Danlos syndrome is the type IV vascular or ecchymotic type; patients with this form of the syndrome rarely survive past 20 years of age.
Restriction fragment length polymorphisms for the type III collagen gene have been described.77 Such diagnostic testing has allowed identification of milder variants of this type. Type III Ehlers-Danlos syndrome with benign hypermobility is inherited as an autosomal dominant trait and is one of the types in which secondary OA can become prominent. The biochemical defect is not known. Type I has large, irregular collagen fibers by electron microscopy. It is also inherited as an autosomal dominant trait and has been associated with premature OA.78
Restriction fragment length polymorphisms for the type III collagen gene have been described.77 Such diagnostic testing has allowed identification of milder variants of this type. Type III Ehlers-Danlos syndrome with benign hypermobility is inherited as an autosomal dominant trait and is one of the types in which secondary OA can become prominent. The biochemical defect is not known. Type I has large, irregular collagen fibers by electron microscopy. It is also inherited as an autosomal dominant trait and has been associated with premature OA.78
 Figure 13-7 Joint hypermobility in Ehlers-Danlos syndrome. |
Quantification of the degree of hypermobility has been described.79 A typical collapsing skeletal structure on the initial handshake may be a clue.
The development of OA seems to be directly related to the severity of hypermobility and the frequency and degree of trauma to which any given joint is exposed.80 OA associated with Ehlers-Danlos syndrome or other hypermobility has been reported in the hands, knees, ankles, and shoulders. It often appears before the age of 40 years, but not all reviews can confirm an association with OA.81 Beighton80 reported finding no cases of OA of the hip associated with the Ehlers-Danlos syndrome.
Synovial effusions studied have had few cells. Synovial biopsy specimens have shown no distinctive changes by light microscopy.82 Pathologic studies of articular cartilage have not been reported. Abnormal bone morphology has been suggested.83 Radiographs of joints have no unique features, and subluxations, when correctable, may not be appreciated on films.
Mechanisms of production of the OA are suspected to include abnormal cartilage structure or wear caused by excessive motion and inadequate protection from trauma. Whether structural abnormalities related to abnormal collagen occur in capsule and cartilage is not yet known. Studies based on pressure-volume relationships in the knee during distention showed no definite evidence of altered collagen functional properties.84
In addition to OA, joints may be involved by dislocations, instability, noninflammatory effusions, and spinal deformities (kyphoscoliosis).82,85 Other potentially confusing and complicating problems include fibromyalgia,81 increased muscle cramps, and spasm; peripheral circulatory disease, especially in type IV Ehlers-Danlos syndrome; congenital abnormalities of bones; and periarticular hemorrhage.
Treatment in symptomatic patients should include educational efforts to help avoid activities that hyperextend joints. Swimming can be used to strengthen muscles to try to aid joint support. Surgery may be made difficult by unpredictable increased bleeding and poor wound healing, but total knee arthroplasties have been performed successfully.86
Isolated joint hypermobility without Ehlers-Danlos syndrome also seems to be associated with an increased incidence of OA in some reports and, in one study,87 with chondrocalcinosis. These were not prospective studies, so the exact reasons for such relationships are not clear.88 Scott and associates88 found more OA in patients with mild idiopathic joint hypermobility than in age-matched control subjects. The neck, thumb, and knee are involved with OA in patients before the age of 30 years.79 In other studies, no correlation could be found between “benign hypermobility” and OA, other arthritis, or arthralgias.89,90 Kraus et al.91 actually found a protective effect of hypermobility on hand OA. It has been proposed that occupational stresses and hypermobility have an additive effect in causing cases of intercarpal OA.92 Recurrent subluxation of the patella can lead to patellofemoral OA, but Crosby and Insall93 actually found that OA was more common in patients in their series after attempted surgical realignment to prevent dislocation.
Other causes of hypermobility that have been described include Larsen syndrome85 (a congenital condition characterized by depressed bridge of the nose and other altered facial features); Desbuquois syndrome, with prominent eyes and a variety of hand problems; spondyloepimetaphyseal dysplasia; the occipital horn syndrome caused by copper deficiency (formerly type IX Ehlers-Danlos syndrome) or the similar Menkes syndrome; acromegaly; Marfan syndrome; Jaccoud arthropathy after rheumatic fever, in systemic lupus erythematosus, or in KID (keratitis, ichthyosis, and deafness) syndrome72; hyperparathyroidism; hereditary osteochondrodysplasia; progressive arthroophthalmopathy97; Wilson disease; and Noonan syndrome.95 Interestingly, OA does not develop in all of these patients, despite the hypermobility.
Endocrine Diseases
Acromegaly
A growth hormone-secreting tumor of the anterior pituitary in adults leads to slowly progressive overgrowth of soft tissue, bone, and cartilage. Because linear growth is not possible at this time, enlargement is prominent in the acral parts, with gradually increasing size of the hands and feet as well as of the nose and mandible. There is typical coarsening of features. Patients with acromegaly often have increased sweating and moist, thick skin. Mild glucose
intolerance occurs in 50% of patients, because hyperexcretion of growth hormone causes insulin resistance. Diagnosis is based on clinical findings together with laboratory confirmation by demonstration of elevated levels of insulin-like growth factor 1 (somatomedin C) or serum growth hormone levels and failure of suppression of growth hormone with glucose. The severity of acromegaly does not correlate directly with growth hormone levels, probably at least partly because growth hormone effects are mediated indirectly through somatomedins produced in the liver.96
intolerance occurs in 50% of patients, because hyperexcretion of growth hormone causes insulin resistance. Diagnosis is based on clinical findings together with laboratory confirmation by demonstration of elevated levels of insulin-like growth factor 1 (somatomedin C) or serum growth hormone levels and failure of suppression of growth hormone with glucose. The severity of acromegaly does not correlate directly with growth hormone levels, probably at least partly because growth hormone effects are mediated indirectly through somatomedins produced in the liver.96
Peripheral and spinal OA is common in acromegaly. Peripheral joint symptoms occur in about 60% of acromegalic individuals.97,98,99 The joints most commonly involved have been the knees, hips, shoulders, elbows, and occasionally ankles. Although soft tissue swelling, widened distal phalanx bone tufts, and carpal tunnel syndrome are present in the hands, there is little OA. Hip and knee involvement has been disabling in severe acromegaly. Crepitus is very common. There may be small or, rarely, large effusions and apparent synovial thickening, but acute inflammation has not been reported.
Backache occurs frequently, but back motion (as well as peripheral joint motion) is often normal or increased. This is tentatively attributed to the thickened disks and cartilages plus laxity of acromegalic ligaments.100 A kyphotic posture is common. There can be spinal demineralization.101
Synovial effusions have been noninflammatory, as in other osteoarthritides. Fluids with high leukocyte counts have been seen in our series only in patients who also have rheumatoid arthritis or gout.102 Synovial biopsy specimens have shown only mild villous proliferation, focal increased lining cells, and increased vascularity.98,102
The early increased cartilage thickness producing wide “joint spaces” on radiographs can be seen at various joints. Later, joint space narrowing, osteophytes, and subchondral sclerosis occur. Chondrocalcinosis, capsular calcification, and osteochondromas have occasionally been seen. Remodeling of phalanges can produce thickening of the shaft at the tendon and capsular attachments, but thin metacarpal shafts have also been seen, possibly caused by remodeling. In the spine, large anterior osteophytes and ossification in widened disks and in ligaments can be seen. Increased new bone formation can be similar to that seen in diffuse idiopathic skeletal hyperostosis and may be related to increased somatomedins.103,104 Vertebral bodies often develop anteroposterior elongation.100
Mechanisms involved in the secondary OA appear to include dramatic cartilage overgrowth that produces joint incongruity and abnormal wear. Whether abnormal cartilage composition also contributes to degeneration is not known. Hypermobility might also contribute to cartilage abuse. Hypermobility was severe in seven patients studied by Kellgren and coworkers,100 with actual subluxations in two. Chondrocalcinosis seen on radiographs105 and apatite crystals seen so far mainly in synovial biopsy specimens102 might also contribute to OA by either local mechanical effects in the cartilage or low-grade inflammation. Histologic studies of articular cartilage show hyperplasia and hypertrophy of the columnar and basal zones of chondrocytes. Superficial fibrillation and erosion of cartilage at weight-bearing sites occur with time. Marginal osteophyte formation is often excessive.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


