Mechanisms of Joint Pain
Hans-Georg Schaible
Pain in the musculoskeletal system is frequent and often chronic. In particular, some diseases of the joint are quite prominent causes of pain, namely, osteoarthitis (OA), rheumatoid arthritis (RA), and gout, and joints are major sites of injuries. In addition, joints may be involved in diseases of the vertebral column, in addition to other structures of the deep tissue.
In joint diseases, pain typically occurs during movements and loading of the joint, thus appearing as mechanical hyperalgesia in the deep tissue. In many cases, mechanical hyperalgesia reflects pathological disturbances of the joint and is thus considered nociceptive. However, some patients also suffer from persistent pain, for example, night pain in advanced OA, and in these cases, a neuropathic component may contribute to the pain (for review see [13]).
In recent years, research into the mechanisms of the clinical features of joint pain has been intensified, and both experimental and clinical studies are now contributing to a better understanding of joint pain. It has become apparent that chronic joint pain is characterized by neuronal processes at all levels of the neural axis (see Fig. 1-1). Furthermore, there is some progress in the exploration of specific pain mechanisms of different joint diseases such as RA and OA. This chapter will focus on the role of cytokines in the generation of joint pain.
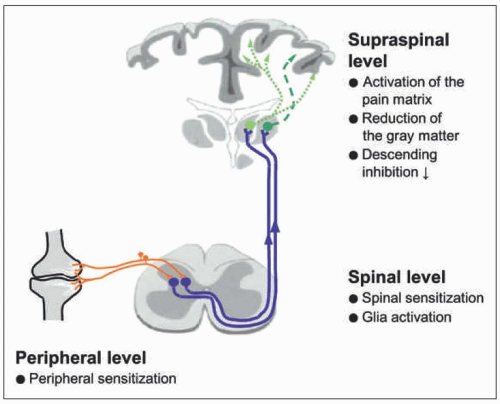 FIGURE 1-1 Overview of neuronal mechanisms that occur during joint diseases. |
Fig. 1-1 shows an overview of neuronal mechanisms that occur during joint diseases. At the peripheral level, joint nociceptors are in a sensitized state. The nociceptive sensory fibers show enhanced responses to noxious stimuli and pronounced responses to innocuous stimuli such as movements in the working range and local pressure onto the joint [16]. Peripheral sensitization is a hallmark of all joint diseases. Because some dorsal root ganglion (DRG) neurons show the expression of ATF3 in the monoiodoacetate model of OA, an additional neuropathic pain mechanism was proposed for OA pain [13].
At the spinal level, nociceptive neurons with joint input are often in a state of central sensitization. Sensitized spinal cord neurons exhibit enhanced responses to innocuous and noxious pressure applied to the joint as well as to stimuli applied to adjacent and even remote regions of the leg [16]. The enhanced responsiveness of spinal cord neurons
corresponds to the spreading of pain in patients suffering from RA and OA [1, 3, 11] (see Fig. 1-2). The generation of spinal hyperexcitability results from an increase in the synaptic processing between sensory neurons and spinal cord neurons. In addition, glial cells may be activated and contribute to central sensitization [9].
corresponds to the spreading of pain in patients suffering from RA and OA [1, 3, 11] (see Fig. 1-2). The generation of spinal hyperexcitability results from an increase in the synaptic processing between sensory neurons and spinal cord neurons. In addition, glial cells may be activated and contribute to central sensitization [9].
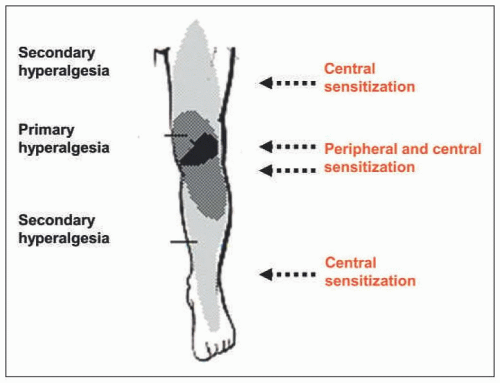 FIGURE 1-2 Areas of primary and secondary hyperalgesia upon inflammation in the knee joint and mechanisms involved. |
In the brain, areas involved in the processing of pain are activated. In addition, structural changes were reported. As in other chronic pain states, a reduction of the gray matter was observed in patients suffering from OA. This process is reversible after replacement of the joint. Strikingly, there may be a loss of descending inhibitory activity in the conditioning pain modulation [1, 3, 13]. The reduction of inhibition is thought to contribute to the sensitized state. The loss of conditioning is also reversible after joint replacement [13].
Recent publications on pain in RA patients and OA patients indicate that the contribution of peripheral and central components to the subjective pain experience may vary. For example, the pain may appear preferentially “peripheral” if typical phenomena of central sensitization such as expanded pain areas and temporal summation are weak. Other patients may appear highly “centralized” if such phenomena are strong [1, 3, 11].
When primary nociceptive neurons are sensitized, both the channels of transduction and the voltage-gated ion channels, in particular the Na+ channels, show changes such that the excitability is enhanced [15]. Numerous mediators have the potential to change the responsiveness of sensory neurons, by acting on second messenger systems that change the activation characteristics of ion channels. While earlier research concentrated on mediators such as bradykinin or prostaglandins, more recent research has focused on mediators such as nerve growth factor (NGF) and cytokines. For the role of NGF in the joint, see [2].
It is usually thought that the neutralization of a proinflammatory cytokine attenuates the disease process and as a consequence, the pain is reduced. Careful observation on experimental models and in patients showed, however, that neutralization of a cytokine may reduce the pain quite quickly, well before the attenuation of the disease can be documented [14]. These observations suggest that certain cytokines have a direct role in the generation and maintenance of pain, that is, by targeting the nociceptive system itself. For several proinflammatory cytokines, direct effects on nociceptive neurons were shown: (a) proportions of nociceptive (and other) sensory neurons express receptors for cytokines; (b) in cultured isolated sensory neurons, the application of cytokines may activate second messenger systems, change the excitability, modify ion currents, and regulate molecules involved in nociception; (c) the injection of some cytokines into normal tissue evokes pain behavior in awake animals and enhances the responsiveness of nociceptive sensory fibers; (d) the neutralization of cytokines may reduce pain well in advance of the attenuation of the inflammatory process [14]. Thus, cytokines contribute to pain through an indirect way by the generation of inflammation that causes the release of many mediators acting on neurons, for example, prostaglandins, as well as through a direct way by acting themselves on neurons.
Proinflammatory cytokines such as TNF-α, interleukin-6, interleukin-1β, and interleukin-17 induce a persistent state of sensitization in
C fibers of the normal joint (Fig. 1-3). After injection into the normal joint, these cytokines enhance the responsiveness of C fibers to innocuous and noxious movements and local stimulation of the receptive field [14]. However, their effects on Aδ fibers vary. The responses of Aδ fibers of the normal joint are increased by TNF-α, on average unchanged by IL-6 and IL-17, but significantly decreased by IL-1β [14]. Thus, each cytokine has its distinct profile on joint nociceptors. Effects of cytokines may also differ in their reversibility. Effects of TNF-α can be reversed by etanercept. By contrast, sgp130, a molecule that binds to complexes consisting of IL-6 and its soluble receptor (sIL-6R) and thus antagonizes their actions on target cells, prevents the sensitizing effect of IL-6 if applied in a pretreatment protocol, but it does not reverse mechanical hyperexcitability if it is applied once IL-6/sIL-6R complexes have already sensitized the nociceptors [14].
C fibers of the normal joint (Fig. 1-3). After injection into the normal joint, these cytokines enhance the responsiveness of C fibers to innocuous and noxious movements and local stimulation of the receptive field [14]. However, their effects on Aδ fibers vary. The responses of Aδ fibers of the normal joint are increased by TNF-α, on average unchanged by IL-6 and IL-17, but significantly decreased by IL-1β [14]. Thus, each cytokine has its distinct profile on joint nociceptors. Effects of cytokines may also differ in their reversibility. Effects of TNF-α can be reversed by etanercept. By contrast, sgp130, a molecule that binds to complexes consisting of IL-6 and its soluble receptor (sIL-6R) and thus antagonizes their actions on target cells, prevents the sensitizing effect of IL-6 if applied in a pretreatment protocol, but it does not reverse mechanical hyperexcitability if it is applied once IL-6/sIL-6R complexes have already sensitized the nociceptors [14].
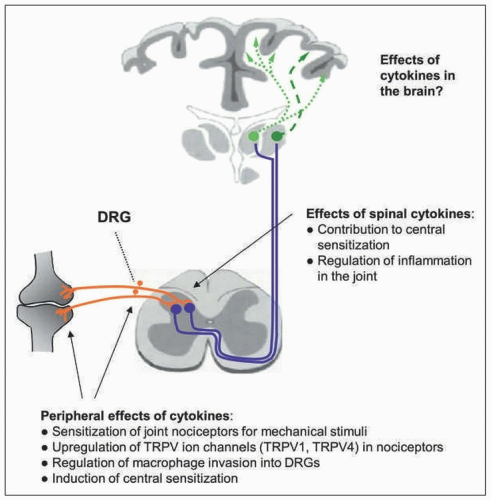 FIGURE 1-3 Effects of cytokines at different neuronal levels. |
The different effects of cytokines are related to the behavioral consequences of cytokine neutralization in the model of unilateral antigen-induced arthritis (AIA) [14]. Neutralization of TNF significantly reduces mechanical and thermal hyperalgesia within 1-2 days although the inflammatory process is barely reduced after such a short time. Such rapid effects of TNF neutralization were also seen in other inflammatory models and in responsive patients with RA. By contrast, neutralization of IL-6 signaling in the AIA model was mainly effective upon pretreatment and only weakly and late upon posttreatment. Neutralization of IL-17 reduced mechanical hyperalgesia but not thermal hyperalgesia, whereas neutralization of IL-1β only reduced thermal hyperalgesia but not mechanical hyperalgesia. The lack of effect of IL-1β neutralization on mechanical hyperalgesia probably results from the contrasting effects of IL-1β on C- and Aδ- fibers [14]. Fig. 1-4 displays the different pattern of cytokine neutralization in the AIA model.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


