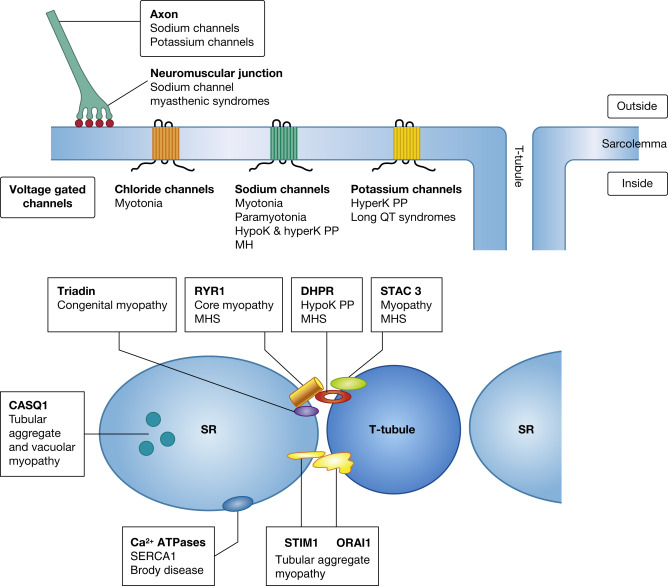
Action potentials in muscle, initiated by depolarization by a nerve impulse and depolarization of the muscle fibre, require the rapid movement of inorganic ions through transmembrane ion channels. The action potential results in the release of calcium ions from the sarcoplasmic reticulum (SR), leading to contraction of the myofibrils. These events of action potential to contraction of muscle fibres are known as excitation–contraction coupling ( ). Mutations in the genes encoding the proteins of these ion channels of the sarcolemma, SR and T tubules, disrupt the normal transport and handling of ions: in particular, sodium, potassium, chloride and calcium ions ( ). They are associated with a number of clinically overlapping syndromes, and defects in the same gene can give rise to varying phenotypes ( Fig. 20.1 , Table 20.1 ). The clinical manifestations of a particular mutation are often determined by its functional effect, which may be either a gain or loss of function. The ion channel proteins are multidomain, transmembrane glycoproteins, and numerous mutations in several genes have now been identified.
| Clinical Syndrome | Type of Ion Channel/Role | Gene | Locus | Inheritance | |
|---|---|---|---|---|---|
| Myotonia congenita (Becker) | Chloride channel | CLCN1 | 7q35 | Recessive | |
| Myotonia congenita (Thomsen) | Chloride channel | CLCN1 | 7q35 | Dominant | |
| Potassium-aggravated myotonia | Sodium channel | SCN4A | 17q23 | Dominant | |
| Paramyotonia congenita | Sodium channel | SCN4A | 17q23 | Dominant | |
| Hypokalaemic or Hyperkalaemic periodic paralysis | Sodium channel | SCN4A | 17q23 | Dominant | |
| Hypokalaemic or hyperkalaemic periodic paralysis | Calcium channel | CACNA1S | 1q32 | Dominant | |
| Episodic ataxia and myokimia | Potassium channel | KCNA1 | 12p13.32 | Dominant | |
| Hyperkalaemic or hypokalaemic periodic paralysis (Andersen–Tawil syndrome) | Potassium channel | KCNJ2 | 17q24 | Dominant | |
| Thyrotoxic periodic paralysis | Potassium channel | KCNJ18 | 17p11 | Unknown/ sporadic | |
| Congenital myopathy | Calcium channel Sodium channel Component of ECC complex Ryanodine receptor Triadin | CACNA1S SCN4A STAC3 RYR1 TRDN | 1q32 17q23 12q13 19q13 6q22.31 | Dominant or recessive Dominant or recessiveRecessive Dominant or recessive Recessive | |
| Congenital myasthenic syndrome | Sodium channel | SCN4A | 17q23 | Recessive | |
| Malignant hyperthermia susceptibilty | Calcium channel Calcium channel Component of ECC complex | RYR1 CACNA1S STAC3 | 19q13 1q32 12q13 | Dominant and ? Recessive Dominant Recessive | |
| Tubular aggregate myopathies | Store operated calcium entry channels | STIM1 ORAI1 | 11p15 12q24 | Dominant Dominant | |
| Tubular aggregates and/or vacuoles | Calcium binding in SR | CASQ1 | 1q23 | Dominant | |
| Brody disease | ATPase calcium pump | ATP2A1 | 16p11 | Recessive | |
There are a number of diverse syndromes involving skeletal muscle which are associated with abnormalities in ion channels. These clinically fall into two broad groups: those with non-dystrophic myotonia or those with periodic paralysis. There have been major advances in the understanding of the molecular basis of many of them, with the identification of defective genes and the interacting proteins that they encode (see Table 20.1 ). In addition, malignant hyperthermia is caused by defects in genes encoding proteins involved with calcium handling (e.g. RYR1, DHPR, and STAC3) and Brody disease by disturbances in a calcium pump of the SR (ATP2A/SERCA1). There are also syndromes affecting the heart (including long QT syndromes and calsequestrin 2-related disorders), various disorders of the central nervous system, such as epilepsy syndromes, ataxias, pain-related disorders, peripheral nerve disorders and neuropathies, and sudden infant death syndromes that are caused by defects in ion channels ( ). In addition, some ion channel defects are associated with myasthenic syndromes (see Ch. 21 ).
Syndromes with Non-Dystrophic Myotonia
Patients with non-dystrophic myotonia typically present with stiffness without weakness or muscle wasting, in contrast to dystrophic myotonic dystrophies (see Ch. 14 ). Molecular defects in genes encoding chloride and sodium ion channels have been identified as causes of these (see Table 20.1 ). Myotonia is a state of delayed relaxation, or sustained contraction, of skeletal muscle. It may manifest after a voluntary muscle contraction, so-called active myotonia , and the patient may be aware of difficulty in relaxing the grip after grasping something. With repetition of the same movement, the myotonia gradually becomes less and then disappears (warm-up phenomenon) and may be accompanied by transient muscle weakness. If the myotonia becomes worse with exercise, instead of improving, the term paradoxical myotonia or paramyotonia is used. In addition, in congenital paramyotonia the muscle stiffness is often profoundly worse with cold environments.
In infants, the first manifestation of myotonia may be delayed opening of the eyes after their closure, with crying, and is specific for dominantly inherited mutations in the skeletal muscle voltage–gated sodium channel encoded by the SCN4A gene. In some patients the myotonia may be fairly localized to only some muscle groups; in others, there may be a more general ‘stiffness’ during the bouts of myotonia.
Myotonia may also be elicited by percussion of a muscle, so-called percussion myotonia . This can usually be demonstrated clinically by percussion with a finger of sites such as the tongue, the thenar eminence, or the deltoid, brachioradialis or gluteal muscles, which give a local contraction dimple.
Both myotonia and paramyotonia show characteristic patterns on electromyograms (EMG) due to repetitive firing of action potentials and imbalances in sodium or chloride ions. With the use of the concentric needle, there is increased activity and irritability of the muscle as the needle is inserted and spontaneous myotonic bursts may be produced. The myotonic bursts can also be elicited if the muscle is tapped with a finger in the vicinity of the needle, or when the patient voluntarily contracts the muscle. These myotonic bursts consist of a prolonged series of rhythmical activity, initially of high frequency (around 20–80 Hz) and high amplitude, and then gradually waning in amplitude and also slowing down. The burst may continue for several seconds and acoustic amplification gives a characteristic sound, likened to a ‘dive-bomber’ or a motorcyclist taking off at speed and disappearing into the distance. The individual elements of the myotonic discharge usually resemble either positive sharp waves or fibrillation potentials and represent action potentials from single muscle fibres.
‘Pseudomyotonia’ is sometimes noted on EMGs in other neuromuscular disorders such as type II glycogenosis. The bursts are usually shorter and less striking than true myotonia and do not show the characteristic decrement. Clinical myotonia cannot be elicited in these patients.
Muscle Pathology
Despite the essential role of ions in muscle function, muscle architecture may not be altered to an appreciable degree. Muscle biopsy is then useful for exclusion of other disorders, and diagnosis relies on thorough clinical and neurophysiological examination.
Many of the pathological descriptions of syndromes now known to relate to ion channel malfunction were reported several years ago before their molecular basis had been determined ( ). The features are non-specific and usually mild ( Fig. 20.2 ). They include variation in fibre size, with atrophy and hypertrophy, and an increase in internal nuclei. In chloride channel myotonias there may be hypertrophy of type 2A fibres, and there may be a reduction in the proportion of this fibre type. In myotonias due to potassium-aggravated sodium channel malfunction, in addition to non-specific variation in fibre size, subsarcolemmal vacuolation relating to dilatation of the T tubules and disruption of myofibrils may be seen with electron microscopy. In paramyotonia congenita, caused by defects in the voltage-gated sodium channel SCN4A gene, pathology is also mild ( Fig. 20.3 ), but tubular aggregates (see below for features) may occur in some cases.


Genotype–Phenotype Correlations of the Myotonias
These disorders are divided into those related to chloride or to sodium channels and include Thomsen and Becker myotonias, potassium-aggravated myotonia and paramyotonia (see Table 20.1 ). Inheritance is generally autosomal dominant, except in Becker myotonia, which is recessively inherited. Thomsen and Becker myotonias are caused by mutations in the gene encoding the voltage-gated chloride channel, CLCN1, on chromosome 7q35. The division into dominant mutations in Thomsen disease and recessive mutations in Becker is now less clear, and a particular mutation can be inherited in either a dominant or recessive pattern ( ). The clinical features associated with both disorders are similar, and both show classical myotonia on EMG and a ‘warm-up’ phenomenon, with improvement on activity. Both can present in childhood, and onset is usually later in Becker myotonia. In Becker, the myotonia is greater in the lower limbs than in the face and arms and may be associated with transient weakness.
Defects in the voltage-gated sodium channel gene ( SCN4A ) on chromosome 17q23 are associated with two clinically overlapping syndromes with myotonia, potassium-aggravated myotonia and paramyotonia congenita . They also cause hyper-and hypokalaemic periodic paralysis syndromes (see below). All these disorders are inherited as an autosomal dominant trait. As the name suggests, potassium ingestion provokes the myotonia in potassium-aggravated myotonia and it may be painful. Paramyotonia congenita shows a characteristic worsening of the muscle stiffness with activity (paradoxical myotonia) and with cold and particularly affects the muscles of the face and hands. On EMG the myotonia is similar to that seen in chloride channel disorders but the effect of cold immersion is an important distinction, with a marked reduction in the evoked compound muscle action potential amplitude.
The mutations in the SCN4A sodium channel gene are distributed throughout the various domains of the channel, but there appears to be a hot spot for paramyotonia congenita in the voltage-sensing region (S4) of domain IV. Many of the mutations lead to a gain in function of the sodium channel, and most lead to impairment of fast inactivation of the channel.
Periodic Paralysis Syndromes
Episodes of weakness or periodic paralysis characterize some disorders associated with disturbances in serum potassium levels, both hypo- and hyperkalaemia or occasionally normal potassium levels ( ). Hypokalaemic periodic paralysis is the most common form caused by mutations in the calcium channel CACNA1S gene in most cases and also by mutations in the sodium channel SCN4A gene ( ). Hyperkalaemic periodic paralysis can also be associated with the sodium channel gene SCN4A and also with potassium channel genes with or without changes in serum potassium ( ; see Table 20.1 ). Andersen–Tawil syndrome is a rare syndrome associated with periodic paralysis, cardiac abnormalities and distinct facial features that are caused by mutations in the potassium channel encoded by the KCNJ2 gene. Thyrotoxic periodic paralysis causes attacks very similar to hypokalaemic periodic paralysis and is associated with defects in the KCNJ18 gene (see Ch. 19 ).
Muscle Pathology
As in non-dystrophic myotonia disorders, periodic paralysis syndromes caused by mutations encoding sodium or potassium channels may show only mild myopathic changes such as variation in fibre size: for example, in Andersen–Tawil syndrome ( Fig. 20.4 ). Other cases, however, may show proliferation and/or dilation of the T-tubular and SR systems. This is identified with light and electron microscopy as vacuoles or as tubular aggregates (see Figure 4.444, Figure 4.445, Figure 4.446, Figure 4.447, Figure 4.448, Figure 4.449, Figure 4.450, Figure 4.451, Figure 4.452, Figure 4.453 and 5.68). The vacuolation is thought to appear in a sequence ranging from focal dilation of the SR to large mature membrane-bound vacuoles ( ). The dilated SR may contain amorphous granular material, cell debris and myelin-like whorls. The vacuolar areas stain for reduced nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR) and acid phosphatase, and immunolabelling shows the presence of dystrophin and β-spectrin, but not laminin, on the membrane ( ).

Tubular aggregates , in addition to staining red with Gomori trichrome, are basophilic with haematoxylin and eosin (H&E) and show reactivity for NADH-TR but not succinate dehydrogenase or cytochrome c oxidase (COX) (see Figs. 4.44 and 20.6). Tubular aggregates are thought to be derived from the SR ( ), but there are examples in which they stain for enzymes thought to be specific for mitochondrial enzymes, such as COX and ubiquinone, and this has not been explained ( ). This might possibly relate to non-specific binding of components of the incubation medium such as nitroblue tetrazolium as tubular aggregates also stain, for example, for myoadenylate deaminase, even without substrate, but see . They also stain for non-specific esterase and other enzymes such as phosphofructokinase. They are often restricted to type 2 fibres and do not show adenosine triphosphatase (ATPase) activity as they contain no myofibrils. With immunohistochemistry, tubular aggregates label with several antibodies, including those to SERCA1 and 2, calsequestrin, ryanodine receptor 1, dihydropyridine receptor, triadin, heat-shock proteins, dysferlin and emerin ( ), but many of the cases reported were not molecularly resolved and had a variety of clinical features.
Tubular aggregates and vacuolar-like pathology not only are associated with periodic paralysis disorders ( ) but also are a particular feature in cases with mutations in STIM1, ORAI1 or CASQ1 (see below). They can also occur in some myasthenic syndromes (see Ch. 21 ), occasionally in cases with phosphoglycerate mutase deficiency and in a variety of other disorders ( ). They may also be seen in paramyotonia congenita caused by defects in the SCN4A gene (see previously).
The voltage-gated potassium channel gene KCNA1 is associated with episodic ataxia and myokymia ( ) but mutations in this gene can also cause a myopathic phenotype with weakness and stiffness and high creatine kinase (CK). Muscle pathology can show variation in fibre size and small type 1 fibres ( Fig. 20.5 ).

Genotype–Phenotype Correlations in Periodic Paralysis
The disorders associated with periodic paralysis fall into two main groups, hyper- and hypokalaemic periodic paralysis : they result from defects in the genes for a sodium ( SCN4A ) or calcium ( CACNA1S ) channel. It will be noted in Table 20.1 that defects in the same genes can be associated with different phenotypes. Hypokalaemic periodic paralysis is the most common form, and mutations in the calcium channel CACNA1S gene that encodes the α1S subunit of the skeletal muscle Ca v 1.1 channel account for about 80% of cases. Approximately 10% of the remaining cases are caused by mutations in the sodium channel SCN4A gene ( ). Whether hyper- and normokalaemic periodic paralysis are distinct entities has been questioned ( ). The episodes of weakness are often triggered by rest after a period of exercise or stress. Hypokalaemic periodic paralysis is characterized by more prolonged (days/weeks) and more severe (four limb) bouts of weakness. Attacks in hyperkalaemic periodic paralysis can last for only a few minutes or hours and may be remarkably focal. They show autosomal dominant inheritance, and onset is usually in childhood or early adolescence.
Patients with hyperkalaemic periodic paralysis overlap clinically with potassium-aggravated myotonia and paramyotonia congenita caused by mutations in the gene for the same sodium channel ( SCN4A ) on chromosome 17q. The same gene can also cause hypokalaemic periodic paralysis. The phenotypic difference is believed to relate to a gain (hyperkalaemic) or loss (hypokalaemic) of function of the ion channel caused by the different mutations. Hypokalaemic periodic paralysis, however, is more commonly caused by mutations in the calcium channel gene CACNA1S on chromosome 1q32. This gene is also associated with malignant hyperthermia (see below).
Mutations in the KCNE3 gene on chromosome 11q13–14 have been described in two families, one with a phenotype consistent with hypokalaemic periodic paralysis, the other with hyperkalaemic periodic paralysis. Recent studies have failed to confirm pathogenicity of the R83H variant and it may be that mutations in KCNE3 modify other ion channel functions rather than cause the condition directly.
have described a syndrome characterized by periodic paralysis, ventricular arrhythmia and mild dysmorphism. The disorder is now referred to as Andersen–Tawil syndrome ( ). Most cases experience hypokalaemic attacks, but hyperkalaemic periodic paralysis has also been described in a minority of cases with this syndrome ( ) and the cause has been shown to be mutations in the KCNJ2 gene on chromosome 17q24 ( ) that encodes the inward rectifying potassium channel Kir 2.1.
Disorders Associated with Calcium Homeostasis
In addition to disorders caused by defective genes that affect chloride, sodium and potassium ion transport, there are several important proteins that control the storage, release and movement of calcium ions. These disorders highlight the importance of store-operated calcium entry and interacting proteins of triads ( ). Genes known to be associated with skeletal muscle disorders that affect calcium homeostasis include those that encode calequestrin-1 ( CASQ1 ), STIM1, ORAI1, RyR1, dihydropyridine receptor (DHPR) and triadin. Triadin is also associated with cardiac defects as well as a skeletal muscle myopathy ( ).
Muscle Pathology
Muscle biopsies from patients with CASQ1, STIM1 or ORAI1 mutations, in addition to tubular aggregates, can also show type 1 fibre predominance, hypotrophic type 2 fibres, internal nuclei and fibrosis ( ). Tubular aggregates are a particular feature of patients with CASQ1 , STIM1 or ORAI1 mutations. As described previously, tubular aggregates stain red with Gomori trichrome, intensely with staining for NADH-TR but negative for succinate dehydrogenase (SDH) and COX ( Figs. 20.6 and 4.44 ). They also stain for myoadenylate and with PAS (periodic acid–Schiff). Tubular aggregates may be restricted to type 2 fibres or be present in both fibre types. With immunohistochemistry, they label with antibodies to SR proteins (see previously), including calsequestrin, sarcoendoplasmic reticulum calcium ATPases SERCAs, STIM1 and RyR1, supporting their origin from the SR. With electron microscopy, the tubules usually show a double membrane (see Fig. 20.6 ). In addition, vacuoles with myelin-like whorls and autophagic debris may be present and, in one case, zebra body–like structures were observed ( ). CASQ1 mutations can also be associated with large vacuoles rather than tubular aggregates ( ). Tubular aggregates have also been observed in individuals with abnormal pupil function and difficulty with night vision (features of Stormorken syndrome), but these cases are molecularly unresolved and may not be caused by an ion channel defect ( ).





