Evaluation of the Patient with Suspected Peripheral Neuropathy
James W. Albers
Introduction
The clinical electromyographer is referred a patient with slowly progressive extremity weakness, mild sensory loss, and areflexia. The referring physician asks only, “Does patient have neuropathy?” After evaluation, possible responses could include “Yes,” “No,” or “I’m not sure,” or the electromyographer could use his or her combined clinical and electrodiagnostic skills to help explain the patient’s symptoms and signs. The evaluation of patients with suspected polyneuropathy or peripheral neuropathy (subsequently referred to as neuropathy) is relatively straightforward. The evaluation consists of a combination of clinical, electrophysiologic, and laboratory studies, with the expectation that the electromyographer will integrate this combined information in arriving at a final diagnosis. It is no longer sufficient to simply confirm the presence of abnormality or to conclude that findings are consistent with neuropathy.
Evaluation of suspected neuropathy is among the most frequent investigations performed in the electromyography (EMG) laboratory, and the electromyographer plays an important role in establishing not only the presence but also the etiology of neuropathy (1). The patient’s history and clinical findings provide important clues in establishing the diagnosis or suggesting studies important in identifying etiology, and the electromyographer’s role in performing a neuromuscular consultation includes paying careful attention to relevant clinical information. Knowledge of potential exposures (occupational, social, or pharmacologic) or recognition of a systemic illness may suggest the cause of a patient’s neuropathy, although symptomatic neuropathy may precede recognition of a systemic disorder. The electromyographer frequently is the clinician most experienced in making such important associations.
The electrodiagnostic examination is simply an extension of the neurologic evaluation. It is derived from sound neurophysiologic principles and provides objective information useful in confirming clinical findings, in addition to localizing abnormalities to a degree not clinically possible. In the evaluation of neuropathy, electrodiagnostic results often suggest the underlying pathophysiology, providing additional clues in establishing etiology. A complete electrodiagnostic study includes evaluation of sensory and motor nerve conduction studies, late responses, and needle EMG.
Almost all patients with neuropathy demonstrate large-diameter fiber dysfunction, making the EMG examination (this term is synonymous with “electrodiagnostic study” mentioned above) a powerful clinical tool for evaluating suspected neuropathy. The presence of large-diameter axon dysfunction is important since these are the nerve fibers most easily accessed by the EMG studies. Classification of neuropathy using electrophysiologic information focuses the differential diagnosis and the subsequent evaluation and often offers a specific diagnosis or class of disorders (2).
Unfortunately, many neuropathies are characterized by nonspecific axonal loss, increasing the importance of other clinical information in establishing a specific diagnosis, which often may include the cause of the neuropathy. Nevertheless, several of the most common treatable neuropathies do have characteristic electrodiagnostic features, yet they were rarely diagnosed until recently. Awareness of these disorders relates to increased use of clinical electrophysiology and identification of characteristic electrodiagnostic features that result in their recognition.
Unfortunately, many neuropathies are characterized by nonspecific axonal loss, increasing the importance of other clinical information in establishing a specific diagnosis, which often may include the cause of the neuropathy. Nevertheless, several of the most common treatable neuropathies do have characteristic electrodiagnostic features, yet they were rarely diagnosed until recently. Awareness of these disorders relates to increased use of clinical electrophysiology and identification of characteristic electrodiagnostic features that result in their recognition.
The following material reviews the underlying pathophysiology associated with neuropathy, defines expectations of the electromyographer, outlines a recommended evaluation, and identifies distinguishing electrophysiologic and clinical features useful in defining specific classes of neuropathy. For each classification, specific clinical examples are included.
Pathophysiologic Features of Neuropathy
Several major pathophysiologic changes are important in the clinical electrodiagnostic evaluation of neuropathy. The most important include axonal degeneration, axonal atrophy, demyelination (uniform and multifocal), and metabolic or ionic channel changes that alter nerve conduction (2,3).
Axonal Lesions
Axonal degeneration results from disorders of the nerve cell (neuronopathy) or axon (axonopathy). Pathophysiologic findings resemble those associated with nerve transection, varying only in degree (3). Separation of the distal axon from the nutritive cell body produces distal axonal degeneration and breakdown of the myelin sheath (Wallerian degeneration). Landau reported that muscle contraction produced by nerve stimulation distal to the lesion persisted for several days after transection, but then disappeared (4). Sensory and motor electrical responses similarly remain normal for several days, with stimulation distal to the transection. Within days, sensory nerve action potential (SNAP) and compound muscle action potential (CMAP) motor amplitudes diminish and ultimately disappear, although conduction along individual axons remains relatively normal before disappearance.
Following nerve transection, the needle EMG examination initially shows absent voluntary activity but no other findings. Gilliatt and Taylor (5) demonstrated spontaneous discharge of individual muscle fibers (i.e., fibrillation potentials) beginning 1 to 4 weeks after axonal degeneration, depending upon proximity to the transection (first appearing in muscles closest to the lesion). Fibrillation potentials reflect muscle fiber hypersensitivity to acetylcholine (ACh) and are associated with proliferation and migration of extrajunctional acetylcholine receptors (AChRs) on the muscle membrane (6). Initial findings include increased muscle fiber sensitivity to mechanical stimulation (typically in the form of positive waves), followed by sustained spontaneous activity (fibrillation potentials) at rest. The amplitude of fibrillation potentials and positive waves diminishes over time, proportional to muscle fiber atrophy, and provides a useful marker for assessing the duration of partial denervation.
Findings with partial or incomplete axonal lesions are similar, with decreased (instead of absent) sensory and motor responses, normal conduction along surviving axons, and reduced voluntary motor unit action potential (MUAP) recruitment (3). Abnormal spontaneous activity appears in denervated muscle fibers. After partial denervation, some denervated muscle fibers eventually are reinnervated by collateral sprouts from surviving axons, resulting in large motor units when the total number of motor units remains reduced (7). ACh hypersensitivity resolves once muscle fibers are reinnervated, and abnormal spontaneous activity disappears. Ballantyne and Hansen demonstrated that regenerating axons produced new motor units by reinnervating muscle fibers shed by abnormally large motor units (8).
The most common morphologic response to a variety of disorders producing neuropathy is a distal axonopathy (3). A variety of mechanisms exist to explain distal axonal degeneration in neuropathy, including failure of axonal transport of some nutrient required for maintenance of the distal axon, as proposed by Schaumburg et al (9). The concept of axonal atrophy is controversial, but it may represent a form of incomplete axonal lesion appearing before axonal degeneration. It is described at the
terminal axon as a reduced diameter of the distal axon (axonal stenosis). Because conduction velocity is proportional to axonal diameter, action potential propagation is reduced proportional to the size of the atrophic distal axon. Unlike axonal degeneration, sensory and motor amplitudes are not substantially reduced in axonal atrophy, and muscle membrane excitability is unaffected.
terminal axon as a reduced diameter of the distal axon (axonal stenosis). Because conduction velocity is proportional to axonal diameter, action potential propagation is reduced proportional to the size of the atrophic distal axon. Unlike axonal degeneration, sensory and motor amplitudes are not substantially reduced in axonal atrophy, and muscle membrane excitability is unaffected.
Myelin Sheath Lesions
Disorders of the myelin sheath produce conduction abnormalities similar to those associated with focal nerve compression, whereby conduction may be slowed or blocked across the site of compression without producing axonal degeneration (10,11). In experimental models using a compressing tourniquet, Ochoa et al identified localized defects beneath the edges of the tourniquet, with telescoping of one myelin segment beneath the next at the node of Ranvier (12,13). This structural abnormality is thought to reduce or block ionic current flow by occluding the node, thereby slowing or preventing action potential propagation (10). Paranodal demyelination is associated with a variety of conditions, including focal compression and neuropathy, and conduction block is attributed to localized demyelination and intramyelin edema. Ochoa and Marotte demonstrated that chronic nerve compression produces similar findings with distorted myelin segments (segmental demyelination), exposed axon membrane, and paranodal remyelination with short internodal distances (11). Membrane excitability does not increase substantially with demyelinating lesions. Conduction slowing or block across a compressive lesion is relevant to the evaluation of generalized neuropathy, in that findings attributed to acquired demyelinating neuropathies have similar myelin abnormalities distributed throughout the peripheral nervous system, with combined demyelination and remyelination, short internodes, and reduced conduction velocity.
Metabolic Lesions
Reduced conduction velocity does not always indicate histologic abnormality such as axonal stenosis or demyelination, because metabolic disorders may produce conduction slowing without identifiable structural abnormalities (3). Among patients with hyperglycemia, increased conduction velocity 6 hours after normalizing glucose levels suggests that nonstructural changes account partially for conduction slowing (14). In association with the neuropathy attributed to chronic diabetes mellitus, the metabolic pathophysiology is poorly understood. Hyperglycemia is believed to produce a decrease in nerve myo-inositol and increased polyol pathway activity related to the increased conversion of glucose to sorbitol by aldose reductase. The reduced nerve myo-inositol leads to reduced Na+/K+-ATPase activity and a resultant increase in intracellular Na+ (15). In isolation, the resultant mild depolarization of the resting membrane potential decreases conduction velocity independent of structural alteration. Additional changes, including inactivation of sodium channels and axoglial disjunction, may also contribute to conduction velocity abnormalities (16). Finally, coexisting microvascular injury to the vasa nervorum and diminished production of nitrous oxide contribute to axonal ischemia and cellular damage, as may possible autoimmune damage mediated by antineuronal antibodies (17).
What is Expected of the Electromyographer?
The electromyographer or electrodiagnostic physician plays an important role in the evaluation of suspected peripheral nerve disorders. It is expected that the electromyographer does more than confirm the presence of abnormality or conclude that findings are consistent with a “neuropathy”; possible etiologies for the neuropathy should be suggested. The electromyographer is a neuromuscular specialist with experience in the evaluation and treatment of patients with neuropathy. It is this clinical experience, combined with electrophysiologic information, that is useful in deriving a diagnosis and establishing the cause of the problem. The emphasis of this chapter is on the application and interpretation of electrodiagnostic information. However, the study begins with a focused history and neurologic examination. Features of the clinical examination particularly important to the evaluation of neuropathy are summarized in Table 11-1.
The electrodiagnostic information is useful only when collected appropriately. Several components
of the examination can be standardized. In the examination, the electromyographer must attend to these components, which are covered in the following series of questions:
of the examination can be standardized. In the examination, the electromyographer must attend to these components, which are covered in the following series of questions:
Table 11-1 Important Features of the Clinical Examination in Suspected Neuropathy | ||
|---|---|---|
|
Were the clinical findings considered in designing the electrodiagnostic evaluation?
Were the limb temperatures monitored and cool limbs warmed to at least 31° to 32°C?
Were the measurement techniques described?
Were normal values provided?
Was the evaluation sufficient to both document the problem and exclude alternative explanations (avoiding errors of omission)?
Were appropriate negative findings discussed?
Was the EMG interpretation consistent with the clinical signs?
Was the referral question(s) adequately addressed?
Although clinical skills are important in documenting the distribution and magnitude of
a suspected neuropathy, the electromyographer’s role is to help identify the underlying pathophysiology and focus the differential diagnosis so that appropriate laboratory investigations can be ordered. Basic questions that should be addressed by the electrodiagnostic study in the evaluation of neuropathy are listed in Table 11-2. These additional questions also extend those addressed by the clinician prior to performing the study.
a suspected neuropathy, the electromyographer’s role is to help identify the underlying pathophysiology and focus the differential diagnosis so that appropriate laboratory investigations can be ordered. Basic questions that should be addressed by the electrodiagnostic study in the evaluation of neuropathy are listed in Table 11-2. These additional questions also extend those addressed by the clinician prior to performing the study.
One of the most important tasks of the electromyographer is to distinguish axonal loss lesions from lesions characterized by uniform or multifocal demyelination. This allows for the possible identification of acquired demyelinating neuropathies, which are important because they are among the most common treatable neuropathies and because they are frequently associated with a systemic illness. The electromyographer also must exclude disorders that mimic neuropathy but are difficult to identify clinically. For example, identifying fibrillation potentials in paraspinal muscles differentiates a distal neuropathy from a polyradiculopathy or a polyradiculoneuropathy. Another example is distinguishing neuropathy from a confluent mononeuritis multiplex. Although clinically difficult, the diagnosis of an underlying vasculitis may be suggested by asymmetric EMG findings, distinguishing a vasculitic neuropathy from other more symmetric forms of distal neuropathy. The answers to the questions in Table 11-2 form the basis of the electrodiagnostic classification of neuropathy, which is described below.
Table 11-2 Expectations for the EMG Evaluation of Neuropathy | ||
|---|---|---|
|
Clinical Electromyography
Nerve conduction studies and needle EMG evaluate slightly different components of the peripheral nervous system. Nerve conduction studies are noninvasive and provide the most useful information in documenting and establishing the type and etiology of neuropathy, whereas the needle EMG examination is more useful in documenting the magnitude and distribution of axonal loss lesions and identifying disorders clinically indistinguishable from neuropathy.
Nerve Conduction Studies
SNAPs and CMAPs are recorded using surface electrodes and percutaneous electrical stimulation (Figs. 11-1 and 11-2) (3). Response amplitudes and latencies are measured and conduction velocities are calculated as part of the evaluation. Conduction over an entire motor nerve is evaluated by F wave latency (Fig. 11-3). F wave measures accentuate mild generalized slowing because of the long conduction distances. Most normal values are age-dependent and some vary according to patient size and other factors (18,19,20,21,22,23). Improper electrode placement, inaccurate measurements, and failure to monitor and control limb temperature influence the results (3). Limb temperature is particularly important in the evaluation of neuropathy. Cooling decreases conduction velocity and increases amplitude, a combination of findings atypical for most pathologic processes. Limb temperature should be monitored, and cool limbs should be warmed to a surface temperature of between 32° and 36°C (23,24). In the context of the electrodiagnostic evaluation of neuropathy, failure to record limb temperature and warm cool limbs limits the ability to interpret the nerve conduction study results.
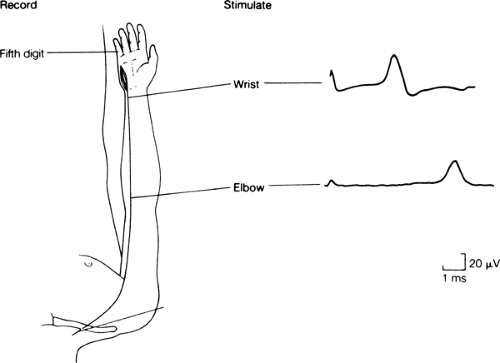 Figure 11-1 • Representative sensory nerve conduction study. Sensory nerve action potentials recorded from the fifth digit following ulnar nerve stimulation at the wrist and elbow. Calibration: 1 ms and 20 μV. (Reprinted from Albers JW, Leonard JA Jr. Nerve conduction and electromyography. In: Crockard A, Hayward R, Hoff JT, eds. Neurosurgery: the scientific basis of clinical practice, 2nd ed. Oxford: Blackwell Scientific Publications Ltd, 1992:735–757 , with permission.) |
Needle Electromyography
The needle EMG examination plays a limited but important role in suspected neuropathy (3). The needle EMG examination evaluates insertional activity (i.e., positive waves and fibrillation potentials) and volitional MUAP recruitment, size, and configuration. As a sensitive indicator of denervation, the needle EMG examination documents the distribution of axonal lesions, providing information from muscles inaccessible to nerve conduction study (e.g., the paraspinal muscles).
Recruitment refers to the sequential introduction of additional MUAPs into the interference pattern as force is increased. In the evaluation of neuropathy, the needle EMG examination provides an indication of ongoing or previous denervation. It also is used to define the distribution of axonal lesions, identifying disorders sometimes confused with or superimposed upon a generalized neuropathy. The amplitude of positive waves or fibrillation potentials and the configuration of MUAPs are used to distinguish acute from chronic disorders and provide an estimate of the rate of progression of axonal loss.
Recruitment refers to the sequential introduction of additional MUAPs into the interference pattern as force is increased. In the evaluation of neuropathy, the needle EMG examination provides an indication of ongoing or previous denervation. It also is used to define the distribution of axonal lesions, identifying disorders sometimes confused with or superimposed upon a generalized neuropathy. The amplitude of positive waves or fibrillation potentials and the configuration of MUAPs are used to distinguish acute from chronic disorders and provide an estimate of the rate of progression of axonal loss.
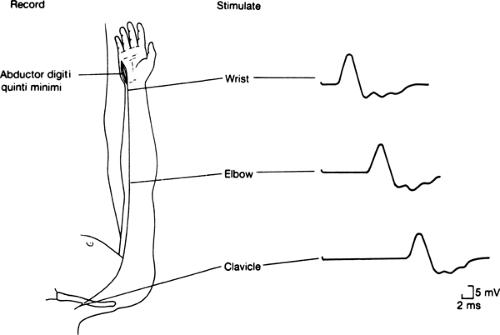 Figure 11-2 • Representative motor nerve conduction study. Compound muscle action potentials recorded from hypothenar muscles following ulnar nerve stimulation at the wrist, elbow, and clavicle. Calibration: 2 ms and 5 mV. (Modified from Albers JW, Leonard JA Jr. Nerve conduction and electromyography. In: Crockard A, Hayward R, Hoff JT, eds. Neurosurgery: the scientific basis of clinical practice, 2nd ed. Oxford: Blackwell Scientific Publications Ltd, 1992:735–757 , with permission.) |
Electrodiagnostic Evaluation in Suspected Neuropathy
Patients commonly are referred for evaluation because of symptoms or signs suggestive of neuropathy. Occasionally, asymptomatic patients are referred because of an underlying illness associated with neuropathy. Some patients referred for other reasons are found to have an unsuspected neuropathy. Regardless of the reason for referral, the initial evaluation for possible neuropathy is based upon the patient’s history and clinical findings. Initial impressions are confirmed or altered, and the study is modified to accept or reject additional considerations until a final diagnosis is achieved.
Protocols for the evaluation of suspected neuropathy are straightforward (Table 11-3). When signs are mild, the evaluation is directed toward the most sensitive or susceptible sites (e.g., the distal lower limb sensory nerves). When severe, evaluation of less-involved sites is important because absent responses provide no information about the presence or absence of conduction slowing. Bilateral studies are performed on some nerves to evaluate symmetry, although a superimposed focal abnormality should not exclude
the diagnosis of neuropathy. The needle EMG examination provides information supplementary to that obtained from the conduction studies. The examination evaluates muscles inaccessible to conduction study, such as paraspinal muscles in suspected radiculopathy, and the results are used to demonstrate a proximal-to-distal abnormality gradient in neuropathy.
the diagnosis of neuropathy. The needle EMG examination provides information supplementary to that obtained from the conduction studies. The examination evaluates muscles inaccessible to conduction study, such as paraspinal muscles in suspected radiculopathy, and the results are used to demonstrate a proximal-to-distal abnormality gradient in neuropathy.
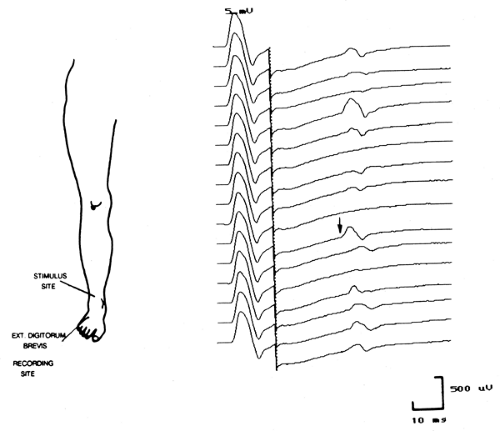 Figure 11-3 • Representative F waves following antidromic peroneal nerve stimulation. (Reprinted with permission from Albers JW. Clinical neurophysiology of generalized polyneuropathy. J Clin Neurophysiol 1992;10:149–166. ) |
Interpretation of Findings
The initial goals are to determine the presence and location of sensory or motor involvement (see Table 11-2). Clinically apparent sensory loss may reflect a lesion proximal to the dorsal root ganglia, whereas abnormal SNAPs document peripheral involvement at or distal to the dorsal ganglia (3). Weakness and atrophy in combination with low CMAP amplitudes reflect abnormality of the lower motor neurons or axons. When present in isolation, these findings cannot localize the lesion more precisely (25,26). The distribution of needle EMG examination abnormalities is helpful in further localizing the abnormality.
The next goal is to identify to the extent possible the primary pathophysiology, such as axonal degeneration or demyelination (3). Axonal neuropathies usually are easily identified. They are characterized by reduced amplitudes, little evidence of conduction slowing, and neurogenic changes on needle EMG with evidence of active denervation and reinnervation (i.e., decreased MUAP recruitment and increased MUAP amplitude, duration, and polyphasia). Using a computerized model of the peripheral nerve, expected CMAP responses for a normal nerve are shown in Figure 11-4. Motor conduction abnormalities associated with axonal degeneration are shown in Figure 11-5. With a loss of
75% of the axons, the CMAP amplitude is markedly diminished, but conduction velocity is reduced only to the extent that is associated with the loss of the largest myelinated axons. There is no evidence of abnormal temporal dispersion.
75% of the axons, the CMAP amplitude is markedly diminished, but conduction velocity is reduced only to the extent that is associated with the loss of the largest myelinated axons. There is no evidence of abnormal temporal dispersion.
Table 11-3 Representative Electrodiagnostic Protocol for Evaluating Polyneuropathy | |||||
|---|---|---|---|---|---|
|
Although primary demyelination is characterized by conduction slowing, overemphasis on mild or focal slowing is a common error in establishing the presence of demyelination (3). Important differences exist between hereditary and acquired demyelination. Hereditary disorders of peripheral myelin (e.g., hereditary motor sensory neuropathy type I) have uniform involvement of all myelinated fibers. Conduction along individual
fibers may be greatly reduced, but slowing is uniform; abnormal temporal dispersion is present only if conduction velocities are markedly slowed and there is substantial phase cancellation. Conduction slowing is disproportionate to the relatively preserved response amplitudes following distal and proximal stimulation (27). Evidence of conduction block is not a typical feature of hereditary demyelination unless conduction velocities are very slow and produce substantial phase cancellation.
fibers may be greatly reduced, but slowing is uniform; abnormal temporal dispersion is present only if conduction velocities are markedly slowed and there is substantial phase cancellation. Conduction slowing is disproportionate to the relatively preserved response amplitudes following distal and proximal stimulation (27). Evidence of conduction block is not a typical feature of hereditary demyelination unless conduction velocities are very slow and produce substantial phase cancellation.
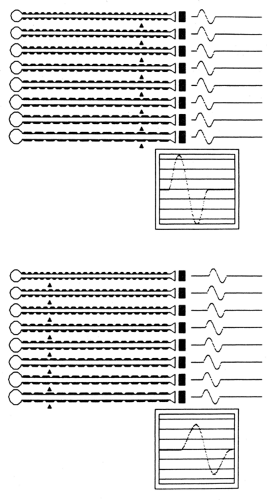 Figure 11-4 • Peripheral motor nerve conduction model demonstrating the resultant compound muscle action potential (CMAP) produced by each of eight individual muscle fiber action potentials and their summation. Individual axons differ in diameter and therefore conduct at different rates. Arrows represent stimulation sites. Top: CMAP (shown in screen) following distal stimulation. Bottom: CMAP following proximal stimulation. (Reprinted from Albers JW. Inflammatory demyelinating polyradiculoneuropathy. In: Brown WF, Bolton CF, eds. Clinical electromyography. Boston: Butterworth, 1987:209–244 , with permission.) |
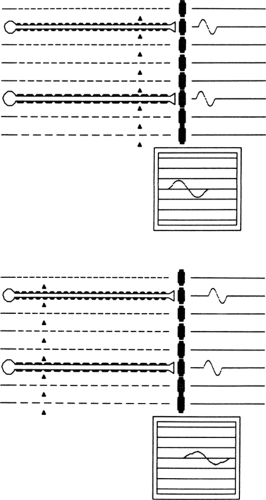 Figure 11-5 • Model of axonal degeneration in motor nerve conduction model described in Figure 11-4, demonstrating CMAPs after random loss of 75% of axons. Arrows represent stimulation sites. CMAPs following distal (upper screen) and proximal (lower screen) stimulation. (Reprinted from Albers JW. Inflammatory demyelinating polyradiculoneuropathy. In: Brown WF, Bolton CF, eds. Clinical electromyography. Boston: Butterworth, 1987:209–244 , with permission.) |
Acquired demyelination is characterized by multifocal, nonuniform abnormalities. The disproportionate involvement of some myelinated fibers compared to others produces increased CMAP duration and a characteristic temporal dispersion of the CMAP (Fig. 11-6) (28). Partial conduction block results from transmission failure along the axon. Because the likelihood of conduction block in any given fiber is length-dependent, there is abnormal dispersion of the response and evidence of partial conduction block when the results of proximal stimulation are compared to those from distal stimulation. This is demonstrated in the model in Figure 11-7 after random, multifocal demyelination, in which propagation is slowed across a single demyelinated internode and blocked if two adjacent internodes are demyelinated (shown by the absence of the myelin sheath). Proximal stimulation produces a CMAP of slightly reduced amplitude and increased duration because of increased dispersion. The area beneath the negative phase of the CMAP is only slightly reduced. Proximal stimulation produces a low-amplitude, highly dispersed CMAP because of the variable amounts of demyelination in some fibers compared to others, producing an increased range of axonal conduction velocities. The initial component of the CMAP is greatly separated from the trailing portion of the CMAP, representing the fastest- and slowest-conducting axons respectively. Phase cancellation of the dispersed responses produces an additional reduction in the proximal CMAP amplitude. Conduction block in two of the axons further reduces the CMAP amplitude to a greater extent than could be explained by abnormal temporal dispersion alone.
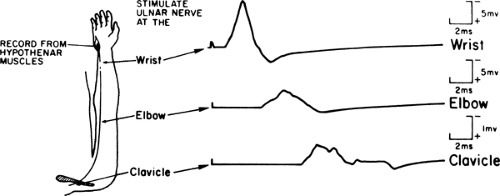 Figure 11-6 • Compound muscle action potentials recorded from hypothenar muscles following ulnar nerve stimulation at distal and proximal sites. Responses are from a patient with an acquired demyelinating neuropathy and demonstrate abnormal temporal dispersion with partial conduction block, increased duration, and decreased conduction velocity. (Reprinted from Albers JW, Kelly JJ Jr. Acquired inflammatory demyelinating polyneuropathies: clinical and electrodiagnostic features. Muscle Nerve 1989;12:435–451 , with permission.) |
Numerous criteria exist to identify acquired demyelination (Table 11-4), but all of the criteria have limitations (28,29,30,31,32). In some criteria, conduction velocity and distal latency thresholds are amplitude dependent. In general, conduction velocities that are less than 70% of the lower limit of the normal range cannot be attributed to axonal loss alone (28). The criteria are not intended to provide strict cutoffs and a “yes-or-no” response. Rather, the results should be used to raise or reduce suspicion for a disorder associated with substantial conduction slowing. The presence of fibrillation potentials and neurogenic MUAP findings does not exclude the diagnosis of demyelinating neuropathy because most hereditary and acquired demyelinating neuropathies have some superimposed axonal degeneration.
The remaining goals of the electrodiagnostic examination are to characterize the neuropathy’s distribution, severity, rate of progression, and
prognosis. The distribution of neuropathy is usually defined clinically, not electrodiagnostically, except for the needle EMG examination of paraspinal muscles in polyradiculopathy or the intrinsic foot muscles in mild axonal neuropathy. Neuropathic severity is best related to CMAP and SNAP amplitudes, because they are proportional to the extent of abnormality, particularly in axonal disorders. The needle EMG examination is useful in documenting very mild axonal neuropathies, as noted above. Defining severity in demyelinating neuropathy is difficult because conduction slowing is not usually associated with functional impairment. Conduction block results in weakness, but conduction block may be difficult to distinguish from abnormal temporal dispersion. Serial electrophysiologic studies are used to estimate the rate of progression, although a mixture of large- and small-amplitude fibrillation potentials can suggest active and chronic denervation.
prognosis. The distribution of neuropathy is usually defined clinically, not electrodiagnostically, except for the needle EMG examination of paraspinal muscles in polyradiculopathy or the intrinsic foot muscles in mild axonal neuropathy. Neuropathic severity is best related to CMAP and SNAP amplitudes, because they are proportional to the extent of abnormality, particularly in axonal disorders. The needle EMG examination is useful in documenting very mild axonal neuropathies, as noted above. Defining severity in demyelinating neuropathy is difficult because conduction slowing is not usually associated with functional impairment. Conduction block results in weakness, but conduction block may be difficult to distinguish from abnormal temporal dispersion. Serial electrophysiologic studies are used to estimate the rate of progression, although a mixture of large- and small-amplitude fibrillation potentials can suggest active and chronic denervation.
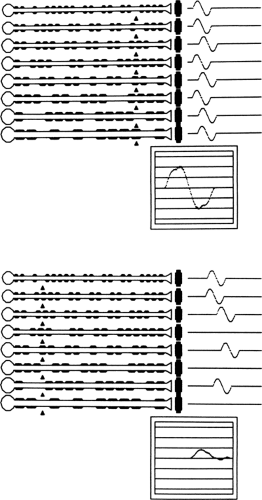 Figure 11-7 • Model of focal demyelination in motor nerve conduction model described in Figure 11-4. Arrows represent stimulation sites. CMAPs following distal (upper screen) and proximal (lower screen) stimulation. The reduced amplitude with proximal stimulation reflects increased temporal dispersion and conduction block in some axons where the individual responses are absent. (Reprinted from Albers JW. Inflammatory demyelinating polyradiculoneuropathy. In: Brown WF, Bolton CF, eds. Clinical electromyography. Boston: Butterworth, 1987:209–244 , with permission.) |
Electrodiagnostic Classification of Neuropathy
Neuropathy is classified by a variety of means, including clinical, biochemical, pathologic, electrodiagnostic, or a combination thereof. The electrodiagnostic results provide information additional to that obtained from the clinical evaluation and are used to assign patients with suspected neuropathy to general categories, thereby directing the subsequent clinical and laboratory evaluations. The classification that follows separates these disorders into broad categories based on electrodiagnostic evidence of sensory or motor involvement combined with conduction slowing suggesting the presence of uniform or multifocal demyelination versus pure axonal loss lesions (2). This classification scheme is not inclusive and there is substantial overlap between categories. This overlap will be manifest by several forms of neuropathy appearing in more than one of the tables that follow. There also is some subjective component to the categorization scheme, requiring that the electromyographer use substantial clinical common sense. Nevertheless, when combined with the clinical history and examination, the electrodiagnostic results may suggest a specific diagnosis or direct the subsequent evaluation. Selected examples are provided in each category; a more extensive discussion exists elsewhere (2).
One issue not addressed in this classification scheme is symmetry. Most forms of peripheral neuropathy are relatively symmetric. There are exceptions, however, as noted earlier. The most common exceptions involve the different forms of
vasculitis producing multifocal abnormalities that can become confluent. Other examples of asymmetric diseases include multifocal motor neuropathy, hereditary neuropathy with liability to pressure palsy (HNLPP), and the neuropathies associated with dapsone, porphyria, leprosy, sarcoidosis, eosinophilia syndromes, and sensory ganglionopathies.
vasculitis producing multifocal abnormalities that can become confluent. Other examples of asymmetric diseases include multifocal motor neuropathy, hereditary neuropathy with liability to pressure palsy (HNLPP), and the neuropathies associated with dapsone, porphyria, leprosy, sarcoidosis, eosinophilia syndromes, and sensory ganglionopathies.
Table 11-4 Electrodiagnostic Criteria Suggestive of Chronic Acquired Demyelination | ||
|---|---|---|
|
Motor Greater Than Sensory Neuropathy, Uniform Conduction Slowing (Table 11-5)
CASE 1
A 23-year-old man presents with a several-year history of progressive ankle weakness and clumsiness. Examination reveals that he has distal weakness of his upper and lower extremities with footdrop, hammertoe deformities, high arches, mild sensory loss to vibration and touch, areflexia, and large firm nerves. Nerve conduction and needle EMG results, shown in Table 11-6, are characterized by reduced CMAP and SNAP amplitudes, prolonged distal and F wave latencies, and substantially reduced conduction velocities (well below 50% of the lower limit of normal). Initial findings of prolonged distal latency that could have been explained by distal entrapment were excluded by demonstrating similar abnormalities in all the nerves examined. There was little change in the configuration (amplitude, duration, or shape) of the CMAP with distal and proximal stimulation (i.e., there was no evidence of abnormal temporal dispersion or partial conduction block). Chronic neurogenic changes were recorded on needle EMG examination of distal extremity muscles. The findings provide electrodiagnostic evidence of a moderately severe sensorimotor neuropathy of the demyelinating type, with mild superimposed axonal degeneration. The markedly reduced conduction velocity without evidence of abnormal temporal dispersion or partial conduction block is most consistent with a hereditary neuropathy.
The classic example of a neuropathy characterized by a uniform peripheral myelinopathy is hereditary motor sensory neuropathy type I (HMSN I), which is the demyelinating form of Charcot-Marie-Tooth disease. HMSN I is a dominantly inherited hypertrophic neuropathy that presents with the insidious onset of distal weakness and sensory loss in young adult life. The diagnosis is clinically suggested by findings of enlarged nerves, distal weakness with hammertoes and pes cavus, abnormal vibratory sensation, and hyporeflexia (33).
The characteristic electrodiagnostic finding associated with HMSN I is markedly reduced conduction velocity in all tested nerves, often as low as 25 m/s or less (33,34). Conduction velocities typically are below 70% of the lower limit of normal and therefore cannot be explained by axonal loss alone. Abnormal temporal dispersion and partial conduction block usually are not present because of the uniform involvement of the myelin of all axons. Nevertheless, when conduction velocities are extremely slow, phase cancellation may result in findings suggestive of abnormal temporal dispersion (27) (see also Chapter 3). F waves are present unless the CMAP amplitude is very reduced, and latencies are prolonged proportional to conduction velocity slowing. Sensory nerves demonstrate similar conduction abnormalities, but low-amplitude responses often preclude conduction velocity measurement. Most patients show at least some degree of axonal loss, and it sometimes is severe. Needle EMG demonstrates decreased MUAP recruitment proportional to weakness, fibrillation potentials and positive waves, and increased MUAP amplitude and duration reflecting chronic reinnervation. Abnormalities are most prominent distally. The patient presented was found to have an asymptomatic sibling with mild ankle weakness, absent ankle reflexes, equivocal sensory loss, and definite slowing of motor conduction velocities.
Table 11-5 Motor Greater Than Sensory Neuropathy, Uniform Conduction Slowing | ||
|---|---|---|
|
Disorders other than HMSN I, many of which also are familial, present with similar electrodiagnostic findings (35). Most neuropathies resembling HMSN I, such as congenital hypomyelinating neuropathy, Dejerine-Sottas disease, and metachromatic leukodystrophy, are associated with disorders of peripheral myelin. Several disorders are not associated with primary myelin abnormalities but reflect selective loss of large myelinated fiber with preservation of smaller myelinated axons. One example is the neuropathy attributed to amiodarone (36,37). Amiodarone is associated with a slowly progressive motor neuropathy with prominent conduction slowing, often in the range of 20 to 30 m/s. Abnormal temporal dispersion and partial conduction block are not features of this neuropathy, and slowing reflects preferential loss of the largest myelinated fibers. Other medications, including the immunosuppressant tacrolimus (FK506) (38) and the antineoplastic suramin (39), produce a motor neuropathy associated with conduction slowing. The slowing of conduction velocity associated with FK506 may be sufficient to resemble an acquired demyelinating neuropathy (40,41). Animal models of suramin-induced neuropathy suggest the presence of a length-, dose-, and time-dependent sensorimotor neuropathy of the axonal type (42). However, 15% of patients enrolled in a phase I study of suramin developed a neuropathy with clinical and electrophysiologic features suggestive of a Guillain-Barré syndrome (GBS) (43).
Table 11-6 Case 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


