Etiopathogenesis of Osteoarthritis
A. Robin Poole
Farshid Guilak
Steven B. Abramson
Osteoarthritis (OA) is a condition that represents a pathological imbalance of degradative and reparative processes involving the whole joint and its component parts, with secondary inflammatory changes, particularly in the synovium, but also in the articular cartilage itself (Fig. 2-1). Idiopathic primary OA may involve one particular joint, or it may be generalized or involve multiple joints in erosive inflammatory forms1,2 (Table 2-1). The presentation of this pathological condition in joints may be a consequence of the biomechanics within the joint which reveal otherwise masked systemic genetically determined changes. The mechanical pressures within the joint may therefore reveal weaknesses in tissue maintenance that are more widespread than previously considered.
Most forms of OA fall into two categories, depending on the predominant background: those that are primary, and often idiopathic, with abnormalities of joint biomaterial and biomechanically faulty joint structure that may result from a recognizable mutation, and those that are secondary and result from superimposed risk factors affecting distribution and severity of loading forces acting on specific joints, such as joint injury.
Risk Factors for Osteoarthritis
Risk Factors Associated with Abnormalities in Joint Biomaterial and Biomechanical Properties, Chemical Injury, or Endocrine Disorders
Familial OA (Table 2-1) may result from abnormal cartilage structure and properties such as a consequence of a mutation in the type II collagen COL2A1 gene (localized on chromosome 12), which causes not only cartilage dysplasia but also a severe form of OA with defective collagen.3,4 Onset is usually soon after cessation of growth. (See Chapter 6 for other gene defects relevant to OA associated with dysplasias and joint laxity.) Chronic abnormalities of growth plate development and bone growth leading to altered congruity of articulating surfaces can also cause OA. OA can develop in patients after traumatic injury or damage to chondrocytes associated with abnormal deposits in the cartilage matrix found in metabolic diseases such as hemochromatosis, ochronosis or alkaptonuria, Wilson disease, and Gaucher disease. OA can also result from disturbances in cartilage metabolism caused by endocrine disorders, such as acromegaly5 (see Chapter 13).
Risk Factors Related to Abnormalities in the Quality and Distribution of Loading Forces on a Joint
Cartilage damage due to trauma, impact injuries, abnormal joint loading, and excessive wear, or as part of an aging process, can lead to changes in the composition, structure, and material properties of the tissue (Fig. 2-2). These alterations can compromise the ability of cartilage to function and survive in the strenuous mechanical environment normally found in load-bearing joints. Joint injury and subsequent joint instability, from loss of ligamental or meniscal support, are significant risk factors for OA.6 Injury and abnormal loading, due to overexercise or abnormal joint use, are a risk for OA.7,8 In regard to underloading, disuse immobilization from chronic inanition, neurologic disorders, or postoperative casting can lead to cartilage disuse atrophy, with increased vulnerability to cartilage injury, unless exercise programs for rehabilitation are carefully controlled. Evidence for this is available from studies of casted animals and those subsequently subjected to controlled or excessive treadmill exercise. Immobilized joint cartilage exhibits arrest of cartilage proteoglycan aggrecan
synthesis,8,9 altered biomechanical properties,10,11,12 and associated elevated metalloproteinases that may contribute to cartilage damage13(reviewed in Helminen et al.14). Deep ulcerative lesions with elevated protease activity can result from treadmill overexercise after disuse immobilization.15 Standard levels of exercise may cause site-specific changes in proteoglycan content and cartilage stiffness, although these changes are not believed to be deleterious16 and may potentially have a beneficial effect in the normal joint.17,18,19 Altered joint loading due to instability or injury of the joint is now well known to be a significant risk factor for the onset and progression of OA.8,20,21 Alterations in joint loading, brought about through ligament transection22 or meniscectomy,23 may lead to profound and repeatable changes in joint tissues which mimic changes seen in early human OA, including increased hydration and proteoglycan turnover, and decreased tissue stiffness in tension, compression, and shear.11,24,25,26,27,28 Obesity is a well-defined risk factor involving excessive joint loading29 as well as systemic metabolic changes that include low-grade chronic inflammation.30 In this respect, obesity is regarded as the number one preventable risk factor for OA.19,31 Evolutionary adaptations to the upright posture by humans have redistributed preponderant loading forces to new sites, predisposing to an increased risk for development of OA in hips, knees, bunion joints, and the lumbar spine.32
synthesis,8,9 altered biomechanical properties,10,11,12 and associated elevated metalloproteinases that may contribute to cartilage damage13(reviewed in Helminen et al.14). Deep ulcerative lesions with elevated protease activity can result from treadmill overexercise after disuse immobilization.15 Standard levels of exercise may cause site-specific changes in proteoglycan content and cartilage stiffness, although these changes are not believed to be deleterious16 and may potentially have a beneficial effect in the normal joint.17,18,19 Altered joint loading due to instability or injury of the joint is now well known to be a significant risk factor for the onset and progression of OA.8,20,21 Alterations in joint loading, brought about through ligament transection22 or meniscectomy,23 may lead to profound and repeatable changes in joint tissues which mimic changes seen in early human OA, including increased hydration and proteoglycan turnover, and decreased tissue stiffness in tension, compression, and shear.11,24,25,26,27,28 Obesity is a well-defined risk factor involving excessive joint loading29 as well as systemic metabolic changes that include low-grade chronic inflammation.30 In this respect, obesity is regarded as the number one preventable risk factor for OA.19,31 Evolutionary adaptations to the upright posture by humans have redistributed preponderant loading forces to new sites, predisposing to an increased risk for development of OA in hips, knees, bunion joints, and the lumbar spine.32
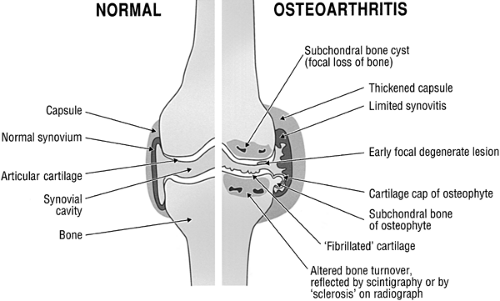 Figure 2-1 Gross pathologic changes observed in OA joints during many years of degenerative change. |
Age-related changes in the magnitude and pattern of stresses on joint cartilage may arise from a number of factors, including altered gait, muscle weakness, changes in proprioception, and changes in body weight. A neuromus-cular control deficit is likely to contribute to the loss of normal attenuation of body weight-bearing forces during walking. This is manifested in OA patients as the “digging of the heels” on forceplate analysis.7 The development of experimental canine OA can be accelerated by posterior nerve root section at the spinal cord, which affects the afferent signals governing stereognostic control of the affected arthritic limb;33 this provides important insights into the pathophysiologic process of the Charcot joint. The mechanism of impact loading, in which energy is normally predominantly absorbed or attenuated by strong muscle groups in the thigh and leg, is also significantly impaired in OA patients,34 who exhibit decreased muscle strength. Quadriceps weakness is associated with knee pain, especially in OA.35 Biomechanical changes in the joint capsule as a consequence of genetic or post-translational changes, such as those affecting collagen fibers, may also influence joint loading.
Examination of nutritional factors in the etiopathology of OA has provided evidence for an increased risk of development of OA of the knee with vitamin E and C deficiencies,36 no doubt influencing matrix assembly and function. In Asia, Kashin-Beck disease, a form of OA, may be caused by dietary exposure to an endemic fungus.37 Hypothyroidism afflicts many of these patients because of a dietary selenium deficiency.38
Articular Cartilage
Composition
Normal articular cartilage consists of an extensive, hydrated extracellular matrix that is synthesized and maintained by a sparse population of specialized cells, the chondrocytes (Fig. 2-3). In the adult human, these cells may occupy as little as 2% of the total volume. The matrix consists mainly of collagen (mostly type II with lesser amounts of other collagen types) and proteoglycans, principally aggrecan, which is large and aggregates with hyaluronic acid (HA).39 Types II, IX, and XI collagens of cartilage combine to form a fibrillar network that provides a structural framework of the matrix in the form of an inhomogeneous and anisotropic meshwork of fibers, which is surrounded by a highly concentrated solution of the proteoglycan aggrecan. A secondary microfibrillar
network involving type VI collagen is concentrated in the pericellular matrix between the cell surface and the territorial matrix.
network involving type VI collagen is concentrated in the pericellular matrix between the cell surface and the territorial matrix.
TABLE 2-1 CLASSIFICATION OF OSTEOARTHRITIS | ||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Proteoglycans are complex macromolecules with a protein core to which are attached glycosaminoglycan chains, primarily of chondroitin sulfate and keratan sulfate. These glycosaminoglycans, which are negatively charged in solution, are responsible for the hydration and large swelling pressure of this tissue.40,41 Many other smaller proteoglycans and other molecules are present within the tissue, some of which may be directly associated with the collagen fibrils. For more details, please see Chapter 4.
Biomechanics of Articular Cartilage
Articular cartilage serves as a low-friction, wear-resistant surface for load support, load transfer, and motion between the bones of the diarthrodial joint. Under normal circumstances, this tissue, bathed in synovial fluid, is able to withstand millions of cycles of loading each year at stresses that may reach 18 MPa,42 while exhibiting little or no wear. These unique properties are endowed by the composition and structure of the major constituents of articular cartilage, and their associated spatial variations provide a complex and inhomogeneous set of material properties.
From a biomechanical standpoint, articular cartilage can be viewed as a fiber-reinforced, porous, and permeable composite material that is saturated with an interstitial fluid, water.43 These characteristics provide for mechanical properties that are viscoelastic (time- or rate-dependent), anisotropic (dependent on direction), and nonlinear (dependent on magnitude of strain). Accordingly, the biomechanical properties of cartilage have been widely studied using models that take into account the multiple phases (collagen solid matrix, interstitial fluid, and mobile and “fixed” ionic groups) and the interactions among these phases.43,44,45
The largest constituent, water, provides load support through fluid pressurization and energy dissipation by fluid flow in response to applied loading.43 The second largest constituent, the collagen fibril, provides articular cartilage with its nonlinear properties in tension,46,47 which are inhomogeneously distributed with depth from the tissue surface. However, these tensile properties decrease progressively with increasing age after 30 years in articular cartilages, such as of the hip, that most commonly develop OA.48 Age-related changes in cartilage tensile properties are believed to arise in part from the accumulation of advanced glycation end products (AGE) which increase the “brittleness” of the cartilage, increasing the potential for tissue fracture and aging-associated biomechanical dysfunction.49
The high concentration of negative charges associated with the proteoglycans in cartilage has a significant effect on the mechanical behavior of the tissue. The negatively charged nature of cartilage arises primarily from the glycosaminoglycans, namely, the many chondroitin sulfate and keratan sulfate chains that are present on aggrecan molecules.50 The large size of aggrecan and its interactions with HA to form macromolecular aggregates serve to retain aggrecan molecules in the extracellular matrix so that their negative charges are “fixed.” These fixed charges are responsible for the physicochemical and electrokinetic properties of this tissue, such as a large osmotic pressure and associated propensity to swell and exhibit streaming potentials, streaming currents, and electro-osmotic effects.41,45 As a result, the mechanical behavior of articular cartilage exhibits an interdependence on physicochemical factors, such as the structural organization and properties
of the collagen fibrillar network, the proteoglycan aggrecan concentration, and the type or distribution of counterions.44 These physicochemical properties significantly contribute to the function of articular cartilage.45 The swelling pressure is believed to largely contribute to residual stresses in the cartilage matrix that enhance the support and distribution of applied loads.51,52 The loss of tensile properties and swelling pressure as collagen and proteoglycans are degraded and lost in OA results in mechanical decompensation and an ensuing increase in the magnitude of tissue strains under similar magnitudes of physiologic loading,10,28,53 particularly at the cellular level.54
of the collagen fibrillar network, the proteoglycan aggrecan concentration, and the type or distribution of counterions.44 These physicochemical properties significantly contribute to the function of articular cartilage.45 The swelling pressure is believed to largely contribute to residual stresses in the cartilage matrix that enhance the support and distribution of applied loads.51,52 The loss of tensile properties and swelling pressure as collagen and proteoglycans are degraded and lost in OA results in mechanical decompensation and an ensuing increase in the magnitude of tissue strains under similar magnitudes of physiologic loading,10,28,53 particularly at the cellular level.54
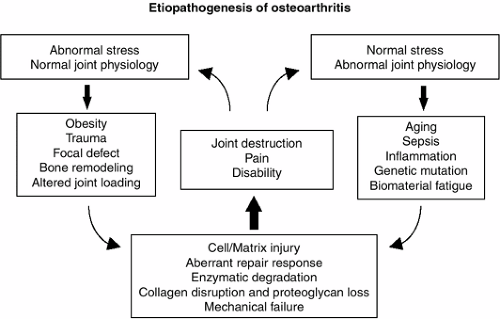 Figure 2-2 Potential mechanisms involved in the etiopathogenesis of OA. |
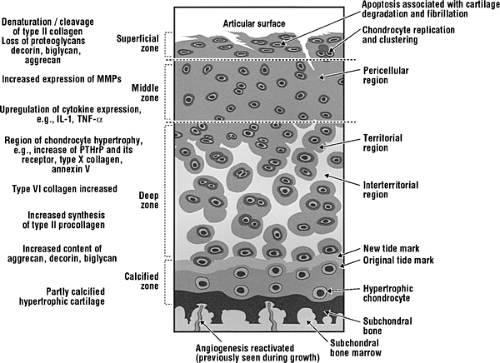 Figure 2-3 Changes observed in articular cartilage in OA involving chondrocytes and extracellular matrix. |
Articular cartilage endows the synovial joint with frictional properties that remain unmatched by man-made joints. The coefficient of friction (ratio of frictional force to compressive force) of cartilage gliding on cartilage has been reported to be as low as 0.002.55 The mechanisms by which the synovial joint achieves these properties involve a combination of biomechanical and biomolecular factors.56
For example, joint congruity and cartilage deformation serve to distribute loads over a larger contact area, thereby minimizing contact stresses. The high water content and low hydraulic permeability of the cartilage extracellular matrix provide a unique mechanism of supporting loads whereby fluid pressurization within the tissue can bear nearly 90% of the load and thus minimize stresses on the solid extracellular matrix.57,58 Synovial fluid plays multiple roles in joint lubrication by providing a high viscosity “squeeze film” layer that delays cartilage-to-cartilage contact under compression59 but also serves as a source of boundary lubricant molecules within the joint.
For example, joint congruity and cartilage deformation serve to distribute loads over a larger contact area, thereby minimizing contact stresses. The high water content and low hydraulic permeability of the cartilage extracellular matrix provide a unique mechanism of supporting loads whereby fluid pressurization within the tissue can bear nearly 90% of the load and thus minimize stresses on the solid extracellular matrix.57,58 Synovial fluid plays multiple roles in joint lubrication by providing a high viscosity “squeeze film” layer that delays cartilage-to-cartilage contact under compression59 but also serves as a source of boundary lubricant molecules within the joint.
Boundary lubricants are molecules that may be adsorbed or bound to the cartilage surface and decrease friction in cases when the tissue surfaces actually contact one another. In the past two decades, there have been significant advances in our understanding of the composition, structure, and properties of biomolecular components of the synovial fluid that serve as the primary boundary lubricants for the joint. “Lubricin,” a ∼ 227 kDa glycoprotein, was first identified from bovine synovial fluid by density gradient sedimentation and gel-permeation chromatography.60 This molecule was first identified as a product of synovial fibroblasts expressing the gene for megakaryocyte stimulating factor (MSF, also termed PRG4). Subsequent investigations have identified a 345 kDa protein, termed “superficial zone protein” (SZP), as an additional product of the MSF gene.61,62 The importance of these molecules in the health of the joint have been demonstrated by the rare autosomal recessive disorder, camptodactyly-arthropathy-coxa vara-pericarditis (CACP) syndrome, which has been attributed to mutations in the PRG4 gene.63 The loss of SZP function in CACP is associated with a severe juvenile-onset joint failure that is noninflammatory in nature.
Biomechanical Regulation of Cartilage Metabolism
A number of in vivo studies have emphasized the relationship between mechanical loading and the health of the joint, and suggest that a critical level and pattern of mechanical stress is required to maintain the normal balance of cartilage synthesis and breakdown. Under normal physiological conditions, the components of the extracellular matrix are in a state of slow turnover, retained in a homeostatic balance between the catabolic and anabolic events of the chondrocytes. These activities are controlled through the processing of both genetic and environmental information, which includes the action of soluble mediators (e.g., growth factors and cytokines), extracellular matrix composition, and physical factors such as mechanical stress. Indeed, many studies have shown that the mechanical stress environment of the joint is an important factor that influences (and presumably regulates) the activity of the chondrocytes in vivo.14,64
Considerable research effort has been directed toward understanding the processes by which physical signals are converted to a biochemical signal by the chondrocyte population. Clarification of the specific signaling mechanisms in normal and inflamed cartilage would not only provide a better understanding of the processes which regulate the physiology of cartilage, but would also be expected to yield new insights on the pathogenesis of OA. In this respect, in vitro explant models of mechanical loading provide a model system in which the biomechanical and biochemical environments can be better controlled as compared to the in vivo situation. Explant models of cartilage loading have been utilized in a number of different loading configurations, including unconfined compression, indentation, tension, shear, and osmotic and hydrostatic pressure (reviewed in Guilak et al.64). The general consensus of these studies is that static compression suppresses matrix biosynthesis, and cyclic and intermittent loading stimulate chondrocyte metabolism (e.g., Gray et al.,65 Sah et al.,66 Guilak et al.,67 Torzilli et al.68). These responses have been reported over a wide range of loading magnitudes, and exhibit a stress-dose dependency. Excessive loading (e.g., high magnitude, long duration) seems to have a deleterious effect, resulting in cell death, tissue disruption, and swelling.69,70,71
The cellular transduction mechanisms responsible for converting mechanical stress into a biochemical response are the subject of much study but are not fully understood. Currently, there is significant evidence that chondrocytes may transduce mechanical signals into biochemical responses through various intracellular and intercellular signaling pathways, including activation of the traditional second messenger pathways such as cyclic AMP (cAMP), inositol trisphosphate, or calcium ion (see Guilak et al.64 and Stockwell72 for reviews).
The Degeneration of Articular Cartilage in Osteoarthritis
The classic loss of articular cartilage observed in OA may be initiated as a focal process that first appears to manifest at the articular cartilage surface. These lesions, which are commonly observed in aging human populations, have all the biochemical features of OA cartilages at arthroplasty.73 They may progressively enlarge to involve specific compartments, inducing alterations in other articulating surfaces by producing changes in loading. Some of the clues as to whether these focal lesions enlarge may have been revealed by recent studies on these lesions in the ankle versus the knee joint. In knee lesions, the emphasis in the cartilage is on matrix degradation, whereas in the ankle it is on matrix synthesis.39 Moreover, in the ankle the response involving increased matrix turnover is more generalized, whereas in the knee it is associated with the lesion.39,73 These fundamental differences in the response and in the lesions could help explain why knee OA is much more common. More widespread changes in loading after traumatic injury may involve the whole joint cartilage, in which alterations in cartilage matrix turnover are detectable within days, weeks, or months after damage to the anterior cruciate ligament or meniscus.74,75 In particular, joint injuries that involve fracture of the articular surface tend to accelerate more rapidly to a degenerative state of post-traumatic arthritis.76
Cartilage degeneration is first observed at the articular surface in the form of fibrillation (Fig. 2-3). Splits are initially parallel to the articular surface; later, they vertically penetrate the damaged cartilage, eventually reaching subchondral bone. Cell cloning is observed early around the splits but is confined to more superficial sites. Many of these cells have become hypertrophic. Progressive loss of cartilage thickness, starting at the surface, is observed.
In large joints, idiopathic OA is ordinarily believed to be a slow process that may take as long as 20 to 30 years, but it is accelerated in cases of joint injury75,77,78,79 or may present clinically on cessation of growth as in familial OA.80 Degeneration is often more pronounced in the tibial cartilage, particularly in the medial compartment. This characteristic is observed in both human and in experimental OA.
Other examples of long-term development of OA come from experimental studies of anterior cruciate ligament transection in dogs, in which bone, ligament, and synovial changes are also observed early28,81,82 (see Chapter 5), presenting as an advanced lesion after a period of only 3 or more years. The earliest changes in the unstable knee that can be seen in articular cartilage in injured joints in animals and humans feature cartilage swelling (also called a hypertrophic reaction), with enhanced synthesis of matrix with an increased content of aggrecan.24 This is followed by a phase of increased matrix turnover, with net depletion of principal matrix components. Finally, severe damage to and loss of the collagen network is observed.11,28,75,83,84 The hypertrophic stage, not to be confused with chondrocyte hypertrophy, clearly precedes the occurrence of the lesional stage with its characteristic deep focal loss of cartilage. In the dog, the biomechanical properties correlate well with a reduction in proteoglycan aggregation and the loss of hyaluronan and link protein.84,85 The presence of smaller proteoglycan aggregates in uncovered or unprotected areas of tibial plateau cartilage in normal control subjects, coupled with the presence of elevated protease activity after surgery, may result in part from a shift of load-bearing sites during the development of OA in this model.86,87
Proteinases of Osteoarthritis Cartilage
Metalloproteinases (MMPs) are generally considered to play a principal role in the cleavage of matrix macromolecules, including type II collagen and the cartilage proteoglycan aggrecan. Collagenases cleave type II collagen, which contains a long triple-helical domain, at a specific site approximately three quarters from the amino terminus. This results in unwinding (denaturation) of the α-chains, which are then susceptible to secondary cleavage by collagenases and other MMPs such as stromelysin 1(MMP-3) and the gelatinases MMP-2 and MMP-9. Aggrecan is cleaved by different MMPs and by the aggrecanases (adamolysins) ADAMTS-4 and -5.
Four collagenases may be active in articular cartilage, namely, MMP-1, -8, -13, and -14 (Table 2-2). MMP-13 is the most efficient at cleaving type II collagen.88 It may be involved in type II collagen cleavage in articular cartilage more than any other collagenase.89,90 Overexpression of constitutively active MMP-13 in cartilage induces the early onset of OA in mice.91 MMP-2 and -9 have also been reported to have collagenase activity. Most MMPs are secreted as latent proenzymes and are then activated. MMP-1 and -13, for example, can bind to collagen fibrils where they can be activated. MMP-3 and MMP-14 are involved in activating other MMPs. Like other MMPs, these activators may themselves be activated by plasmin generated from plasminogen by urokinase-type activator produced by chondrocytes or by cysteine proteinases such as cathepsin B,92 or by MT1-MMP (MMP-14), which possesses a furin cleavage site, which means that it is usually activated within the cell before secretion. Cathepsin K, a cysteine proteinase, can also cleave triple-helical collagen but in primary sites different from the primary cleavage produced by collagenases.93 It is expressed by superficial chondrocytes in increased amounts in OA cartilage.94 There is unpublished evidence thatthis proteinase is involved in collagen cleavage in OA articular cartilage in culture (V. Dejica, J.S. Mort, and A.R. Poole, unpublished data).
In OA, there is increased expression and content of various MMPs, including MMP-1, MMP-3, MMP-13, and MMP-28 (epilysin), but not ADAMTS-4 (a disintegrin and metalloproteinase with thrombospondin motifs) or ADAMTS-5, the latter being downregulated.95 Others have observed that increased MMP-13 expression is the most dominant of the collagenases, being stronger in late stage disease when MMP-3 is downregulated; of the aggrecanases, only ADAMTS-5 was clearly expressed.96 This increased expression is first observed at or close to the articular surface very early in the degenerative process.97,98 Similarly there is increased expression of MMP-2, MMP-9, MMP-8,99,100 MMP-11,101 MMP-14,102,103 and matrilysin.104 Plasminogen activator105 and cathepsin B106,107 are also upregulated in human and experimental OA cartilage.
Cleavage and denaturation of type II collagen by collagenases are first observed around chondrocytes in these superficial sites where MMP-1 and -13 are present in increased amounts.97,108,109 This change is also seen with aging but more so in OA. It extends into the territorial and interterritorial matrix and then progressively extends into the mid and deep zones with lesion development.97 Collagenase activity, the denaturation of type II collagen, and the associated loss of type II collagen73,110 are much increased in OA cartilages.73,89,108 These changes in articular cartilage are seen within months of anterior cruciate ligament rupture, which is a major risk factor for OA development in a joint.75,111
In established OA of the knee, the synthesis of type II collagen is simultaneously increased markedly.39,112,113,114 However, these new molecules, as well as resident pre-existing collagen, are the subject of excessive proteolysis.90 The same applies to aggrecan.115
The core protein of aggrecan is cleaved by different proteinases.50 Cleavage sites are common in the interglobular domain between the G1 and G2 domains as well as in the chondroitin sulfate-rich region. In the interglobular domain, two principal cleavage sites have been identified: the aggrecanase site, where proteinases such as cell surface-associated
aggrecanases or ADAMTS-4 and -5 can cleave;116 the MMP cleavage site is a target for multiple MMPs.92 There is evidence for enhanced degradation of aggrecan at both these cleavage sites in cartilage matrix in OA.117
aggrecanases or ADAMTS-4 and -5 can cleave;116 the MMP cleavage site is a target for multiple MMPs.92 There is evidence for enhanced degradation of aggrecan at both these cleavage sites in cartilage matrix in OA.117
TABLE 2-2 EARLY CHANGES IN THE STRUCTURE AND COMPOSITION OF ARTICULAR CARTILAGE IN HUMAN OSTEOARTHRITIS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Of the aggrecanases, ADAMTS-5 has been shown to be the major enzyme responsible for aggrecan loss in experimental murine OA118 and inflammatory joint disease.119 These proteinases are also activated outside the cell, probably at the cell surface.119,120
A variety of cleavage products of aggrecan accumulate in OA cartilages. Early in cartilage degeneration (Mankin score of 1 to 6), the excessive proteolysis leads to a reduction in molecular size of aggrecan fragments that have accumulated with aging as a result of degradation, probably during a period of many years.115 With increased degeneration, the sizes of aggrecan fragments increase.115 This may reflect altered proteolysis, but is more likely the partial degradation of more recently synthesized larger molecules. That new aggrecan synthesis and incorporation occur is reflected by the appearance of epitopes on chondroitin sulfate that are normally only commonly found in actively biosynthetic fetal cartilages.115,121,122,123 Some of these epitopes that increase in OA in synovial fluid, such as the 846 epitope,124,125 are thought to present on newly synthesized molecules that have been released by matrix degradation.126
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


