Cell Biology, Biochemistry, and Molecular Biology of Articular Cartilage in Osteoarthritis
Linda J. Sandell
Dick Heinegard
Thomas M. Hering
Cartilage: Tissue Organization
Articular cartilage is comprised largely of an extracellular matrix synthesized by chondrocytes.1 In synovial joints, the layer of hyaline articular cartilage tissue faces the joint cavity (i.e., the synovial fluid space) on one side and is linked to the subchondral bone plate via a narrow layer of calcified cartilage tissue on the other. The medial femoral articular cartilage of humans is about 2 to 3 mm thick. The organization of the extracellular matrix and the distribution of zones is slightly different in immature versus mature cartilage. In young individuals, the layer of articular cartilage is generally much thicker and unstratified, with chondrocytes being distributed in a more random, isotropic pattern. As the tissue matures, there is a much higher degree of anisotropy with the cells and matrix being arranged into the clearly defined zones. These changes are accompanied by a significant increase in the mechanical competence of the cartilage with improvement in its stiffness and resistance to shearing and compressive forces.2 The zones of articular cartilage (Fig. 4-1) are the superficial (tangential), the middle (transitional), the deep (radial), and calcified cartilage. The material properties of the cartilage at different depths also change because they are determined by the biochemical nature, content, and organization of the matrix macromolecules. The zonal variations in the extracellular matrix must result from metabolic differences among the cells. In experiments where the different zones are isolated and cultured, striking differences are found in terms of their morphology, metabolism, phenotypic stability, and responsiveness to interleukin-1α (IL-1).3
Chondrocyte Cell Biology
Unlike many other tissues in the body, the only cell type found in cartilage is the chondrocyte.4 Chondrocyte morphology varies from rounded or polygonal, to the flattened, discoid shaped cells at the articular surface of joints. The chondrocyte has intracellular features characteristic of a metabolically active cell (Fig. 4-2), due to its role in synthesis and turnover of extracellular matrix components. Generally, chondrocytes occupy about 10% of the tissue volume in articular cartilage. Embyologically, chondrocytes of limb elements are derived from the mesoderm, and those of the facial skeleton from the neural crest. The earliest differentiated chondrocytes arise following mesenchymal condensation at sites of future skeletal elements, a process involving the expression of tissue specific genes, including Sox9, L-Sox5, Sox6, and collagen
type IIA.5 Whereas chondrocytes in articular cartilage persist, those in the epiphyseal growth plates proceed to become terminally differentiated hypertrophic chondrocytes, a cell that facilitates endochondral ossification. One function of chondrocytes is in growth. Chondrocytes within the epiphysial plates achieve tissue growth through proliferation to increase cell number, through matrix production, and through increased cell volume during terminal differentiation. The major function of chondrocytes within supporting cartilages is to maintain the extracellular matrix. A proteomic reference map of human chondrocytes has been published,6 leading to the identification of 93 different intracellular chondrocyte proteins. Of these, 26% are involved in cell organization, 16% in energy, 14% in protein fate, 12% in metabolism, and 12% in cell stress.6
type IIA.5 Whereas chondrocytes in articular cartilage persist, those in the epiphyseal growth plates proceed to become terminally differentiated hypertrophic chondrocytes, a cell that facilitates endochondral ossification. One function of chondrocytes is in growth. Chondrocytes within the epiphysial plates achieve tissue growth through proliferation to increase cell number, through matrix production, and through increased cell volume during terminal differentiation. The major function of chondrocytes within supporting cartilages is to maintain the extracellular matrix. A proteomic reference map of human chondrocytes has been published,6 leading to the identification of 93 different intracellular chondrocyte proteins. Of these, 26% are involved in cell organization, 16% in energy, 14% in protein fate, 12% in metabolism, and 12% in cell stress.6
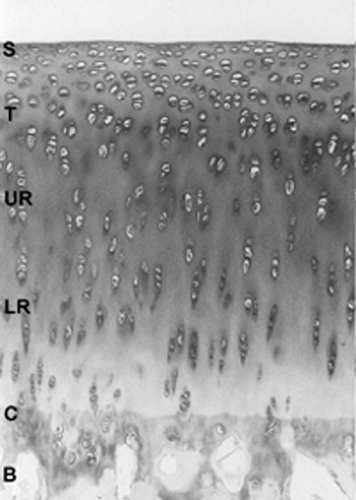 Figure 4-1 Articular cartilage from adult rabbit showing the superficial (S); middle, composed of the transitional (T) and upper radia zones (UR); and deep or lower radial zone (LR). |
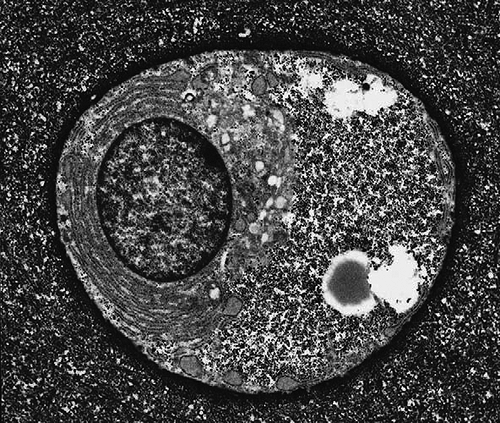 Figure 4-2 Electron micrograph of articular chondrocyte. Cell shape can vary from flattened or discoid near the articular surface, to a more rounded morphology in the middle and deep cartilage regions. Evident in the cytoplasm are cytoplasmic organelles including rough endoplasmic reticulum and Golgi apparatus, as well as glycogen and lipid droplets. Note the high ratio of extracellular matrix to cell volume. (Reprinted from Archer and Francis-West, Int. J. Biochem. Cell Biol 35:401–404, 2003. ) |
Biochemical Composition of the Cartilage Extracellular Matrix
Cartilage is composed of an extracellular matrix that has been biosynthesized by chondrocytes. Compared to other tissues, the ratio of matrix to cell volume is very high. It is a hyperhydrated tissue, with values for water ranging from 60% to almost 80% of the total wet weight. The remaining 20% to 30% of the wet weight of the tissue is principally accounted for by two macromolecular materials: type II collagen, which composes up to 60% of the dry weight, and the large proteoglycan, aggrecan, which accounts for a large part of the remainder. “Minor” amounts of other collagens, including collagen types IX, XI, III, V, VI, X, XII, and XIV, are found in the matrix. Other structural proteins, glycoproteins, proteoglycans, and enzymic molecules make up a small percentage of the tissue, but may be of critical importance in the structure and function of the tissue. Minor amounts of other “large” proteoglycans are found in cartilage including versican, perlecan, and SZP/Lubricin. Among small proteoglycans of the small leucine-rich repeat proteoglycan (SLRP) family, those localized to cartilage include biglycan (DS-PGI), decorin (DS-PGII), epiphycan (DS-PGIII), fibromodulin, and lumican. Additional structural, regulatory, and enzymic proteins of the cartilage extracellular matrix are described below. Figure 4-3 is a schematic representation of many of the molecules of cartilage showing some of their interactions.
The Cartilage Proteoglycan Aggregate
The cartilage proteoglycan aggregate is a unique assembly of macromolecules that, along with type II collagen and a number of accessory molecules, confers upon cartilage its special biomechanical properties. The cartilage proteoglycan aggregate is composed of chondroitin sulfate proteoglycan (aggrecan) monomers bound into very large aggregates with hyaluronan. This interaction is stabilized by the binding of a third component, the link protein, a 45- to 50-kDa glycoprotein which is bivalent, in that it has binding sites
for both the aggrecan amino terminal globular (G1) region and for hyaluronan (Fig. 4-4).
for both the aggrecan amino terminal globular (G1) region and for hyaluronan (Fig. 4-4).
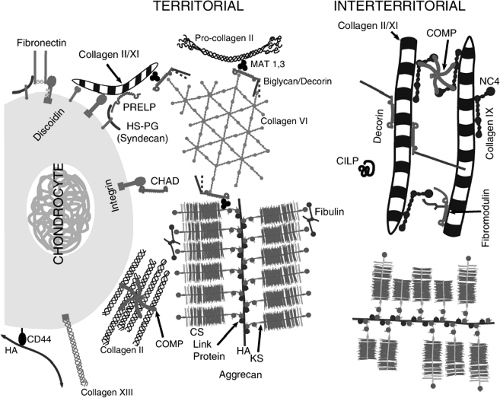 Figure 4-3 Schematic representation of the cartilage matrix. Matrix closest to the chondrocyte is indicated as territorial and between chondrocytes is interterritorial. Note the fibrils of collagen coated with the small proteoglycans decorin and fibromodulin as well as with the collagen IX. The proteoglycans bind by their protein core, leaving their side chains free to self-interact or to interact with other collagen fibers or with the NC4 domain of collagen IX or with fibulin. Proteins and protein interactions are discussed in the text. |
Aggrecan Core Protein Structure
Aggrecan is a large, complex, hybrid proteoglycan having a 220- to 250-kDa multiple-domain core protein which is substituted with both chondroitin sulfate (CS) and keratan sulfate (KS) chains in addition to N- and O-linked oligosaccharides (reviewed in Kiani et al.7). The core protein possesses two globular regions near the amino-terminus, referred to as G1 and G2, separated by an interglobular domain (IGD). A third globular region, G3, is found at the carboxyl terminal end of the core protein. An extended region containing KS- and CS-attachment sites is found between the G2 and G3 domains.
The globular region G1 of aggrecan is composed of three domains referred to as A, B, and B’, and the G2 region is subdivided into B and B’ domains similar to those in G1. The A domain has also been referred to as the “Ig fold” domain due to its sequence homology to immunoglobulin-like proteins8 and has been suggested to mediate the interaction of aggrecan with link protein in the proteoglycan aggregate.9,10 The B and B’ domains of the aggrecan G1 region are believed to mediate interaction with hyaluronan. These domains have also been referred to as proteoglycan tandem repeat (PTR) domains. The related B and B’ (or PTR) domains in the G2 region apparently do not interact with hyaluronan11 and their function is unknown at present.
Between the G2 region and the G3 region of the molecule are found the KS and CS attachment regions. This extended region of the molecule is heavily substituted with KS and CS chains, with KS chains concentrated in the amino-terminal portion of this extended region.12 The KS domain consists mainly of hexapeptide repeats, the number of which is variable between species.13 The CS attachment region is generally subdivided into CS-1, and CS-2 subdomains, which differ in terms of the repeated sequence comprising each subdomain, and in the length and sulfation of the CS chains. Each of these subdomains has been found to be variably conserved between species.14 The number of CS-1 repeats in human aggrecan has been found to be structurally polymorphic, with the number of repeats varying from 13 to 33 between individuals.15,16
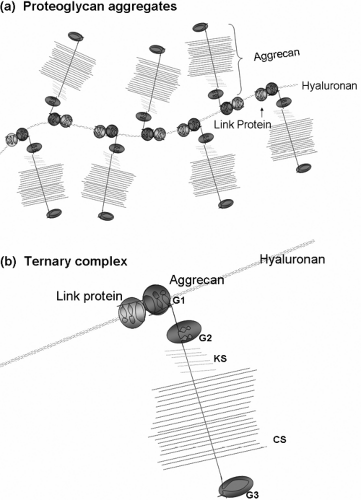 Figure 4-4 The cartilage proteoglycan aggregate is a complex of hyaluronan, aggrecan monomers, and link proteins. (a) Multiple aggrecan monomers bind to a single hyaluronan filament. This interaction is stabilized by link protein. (b) Aggrecan binds to HA and link protein through its G1 domain to form a ternary complex. Highly charged KS and CS polysaccharides are located in the extended region between the G2 and G3 domains of aggrecan. (Reprinted from Miwa HE. A recombinant system to model proteoglycan aggregate interactions and aggrecan degradation. Doctoral dissertation, Case Western Reserve University, Cleveland, OH, 2006. ) |
The G3 domain at the C-terminus of aggrecan is made up of three modules, including an Epidermal growth factor (EGF) like region, a C-type lectin-like domain (also termed carbohydrate-recognition domain [CRD]), and a complement binding protein (CBP)-like domain. The EGF-like region is alternatively spliced in humans and may contain an EGF-1, EGF-2, or both modules.17 The C-type lectin-like module of aggrecan can bind to fucose and galactose.18 In addition, this module is able to bind tenascin-C,18 sulfated glycolipids,19 and fibulin-120 and -2.21
A functionally null mutation of the aggrecan gene has been identified in mice, termed cartilage matrix deficiency (cmd). This is a natural aggrecan gene knockout and was the first example of a proteoglycan gene mutation observed in mammals.22 The cmd aggrecan gene contains a 7 bp deletion in exon 5, which codes for the B loop of the G1 domain, which causes a frame shift and premature termination in exon 6. Homozygotes (cmd/cmd) exhibited dwarfism, a cleft palate, and a short snout and die soon after birth due to respiratory failure. The cartilage of homozygous mice contains tightly packed chondrocytes with little apparent matrix. Heterozygous mice (cmd/+) are born normal. Although cmd mice produce no aggrecan, link protein and collagen type II are produced normally.23
Link Protein
The link protein bears sequence homology and a parallel domain structure to the G1 domain of aggrecan.24 Link protein stabilizes the binding interaction between the hyaluronic acid binding region (HABR) of aggrecan and hyaluronan. Link protein is capable of binding independently to either hyaluronan or the aggrecan G1 domain, and there is evidence that this binding occurs through separate functional domains within the link protein molecule.9 Link protein contains one copy of an immunoglobulin (Ig) fold-like sequence at the N-terminus and also contains two homologous sequences known as PTR domains between the N-terminal Ig fold domain and the carboxyl terminus (Fig. 4-5). The vertebrate hyaluronan and proteoglycan-binding link protein gene family (HAPLN) consists of four members, including cartilage link protein (HAPLN1), two forms that are expressed in the brain and CNS (HAPLN2 and HAPLN4), and a form that is widely expressed (HAPLN3).25
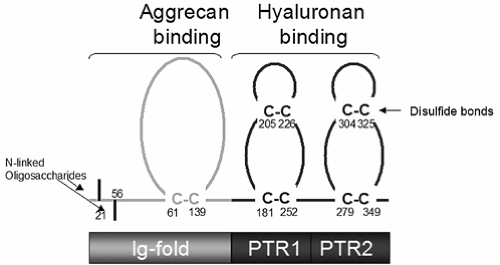 Figure 4-5 Domain structure of cartilage link protein. The N-terminal Ig-fold domain is involved in aggrecan binding and the PTR domains are involved in HA binding. Link protein is a glycoprotein containing N-linked oligosaccharides and multiple disulfide bonds. The structure of the aggrecan G1 domain is closely related to that of link protein. (Reprinted from Miwa HE. A recombinant system to model proteoglycan aggregate interactions and aggrecan degradation. Doctoral dissertation, Case Western Reserve University, Cleveland, OH, 2006. ) |
Homozygous link protein knockout mice (LP-/-)26 exhibit dwarfism and a flat face. As with cmd mice, most homozygotes die shortly after birth due to respiratory failure. Heterozygous mice show no apparent phenotype.
In heterozygotes, long bones are shortened and skulls are small with a shortened anterior-posterior axis. Growth plate in LP-/- mice shows disorganization of the chondrocytes, suggesting that the lack of link protein affects chondrocyte differentiation. As might be expected, the level of aggrecan in LP-/- cartilage is significantly reduced, confirming a role for link protein in aggregate stabilization.
In heterozygotes, long bones are shortened and skulls are small with a shortened anterior-posterior axis. Growth plate in LP-/- mice shows disorganization of the chondrocytes, suggesting that the lack of link protein affects chondrocyte differentiation. As might be expected, the level of aggrecan in LP-/- cartilage is significantly reduced, confirming a role for link protein in aggregate stabilization.
Hyaluronan
Hyaluronan (reviewed in Laurent et al.27) is a polysaccharide having the repeating disaccharide structure poly [(1/3)-β-DGlcNAc-(1/4)-β-D-GlcA-]. Hyaluronan is predominantly localized to the extracellular and pericellular matrix, although it may occur intracellularly.28 Functionally, it contributes to the elastoviscosity of fluid connective tissues29 including synovial fluid and vitreous humor, it modulates hydration and transport of water through tissues, and functions in receptor-mediated cell detachment, mitosis, and migration,30 inflammation,28 tumor development, and metastasis.31 In cartilage, hyaluronan functions in the supramolecular assembly of proteoglycans and link protein into aggregates.1
TABLE 4-1 THE GENETICALLY DISTINCT COLLAGENS IN ARTICULAR CARTILAGE | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Collagens
Type II Collagen
Mature collagen fibers provide the capacity to withstand tensile and shear forces, while proteoglycans are generally responsible for solute flow and deformation of the tissue. The collagens of cartilage are listed in Table 4-1. The predominant collagen in the mature articular cartilage is the fibrillar collagen, type II. Type II collagen is a triple helix composed of three identical alpha chains synthesized from the COL2A1 gene. Type II procollagen is synthesized in two splice forms: type IIA contains an additional cysteine-rich exon in the N-propeptide and is found in chondroprogenitor cells and other embryonic tissues (Fig. 4-6); type IIB lacks this cysteine-rich domain of the N-propeptide and is
the predominant collagen in all cartilaginous tissues. These alpha chains form the characteristic collagen triple helical structure when three chains are wound around each other during biosynthesis to form collagen molecules. The collagen molecules then associate in a lateral staggered array to form collagen fibrils. These fibrils are not necessarily one single type of collagen, even in cartilage, and often contain, within the fibril or surrounding it, other types of collagens. These other collagens are considered “minor” in amount but have important functional roles. Mutations in the predominant collagen, type II, can weaken the cartilage matrix and predispose the tissue to osteoarthritis. The diameter of the collagen fibril in cartilage is shown in Table 4-2. In articular cartilage of mammals, the content and interaction of predominant and minor collagens as well as other collagen-binding molecules contributes to the differences in collagen fibril diameters observed in the different zones of the tissue.
the predominant collagen in all cartilaginous tissues. These alpha chains form the characteristic collagen triple helical structure when three chains are wound around each other during biosynthesis to form collagen molecules. The collagen molecules then associate in a lateral staggered array to form collagen fibrils. These fibrils are not necessarily one single type of collagen, even in cartilage, and often contain, within the fibril or surrounding it, other types of collagens. These other collagens are considered “minor” in amount but have important functional roles. Mutations in the predominant collagen, type II, can weaken the cartilage matrix and predispose the tissue to osteoarthritis. The diameter of the collagen fibril in cartilage is shown in Table 4-2. In articular cartilage of mammals, the content and interaction of predominant and minor collagens as well as other collagen-binding molecules contributes to the differences in collagen fibril diameters observed in the different zones of the tissue.
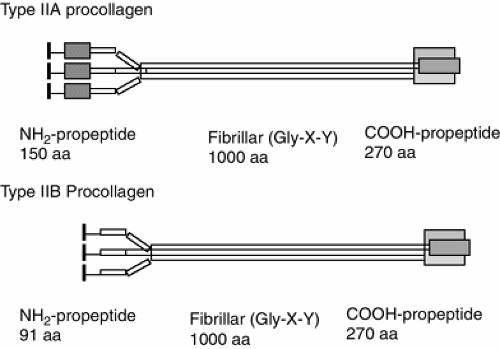 Figure 4-6 Type II collagen. Type IIA procollagen contains an additional cysteine-rich protein domain in the NH2-propeptide (shaded box) that binds to bone morphogenetic proteins during skeletal development. Type IIA procollagen is synthesized by chondroprogenitor cells. Type IIB procollagen is made by chondrocytes and lacks the cysteine rich domain of the NH2-propeptide. In chondroprogenitor cells, Type IIA procollagen is deposited into the matrix with the NH2-propeptide intact. It is subsequently removed by matrix enzymes. In chondrocytes, both propeptides are removed with the mature collagen molecule deposited in the extracellular matrix. This molecule forms associates laterally into the fibrils indicated in Table 4-2. The black box is the signal peptide (removed during translation into protein). |
Type II collagen is biosynthesized as a procollagen molecule containing N- and C-terminal propeptides. In cartilage, the propeptides are removed prior to assembly of the mature collagen. The C-terminal propeptide has been isolated from cartilage (called chondrocalcin) and at one time was thought to be an independent molecule. Although synthesized in all cartilages, chondrocalcin appears to be associated with extracellular matrix undergoing mineralization.32 In chondroprogenitor tissues, type IIA procollagen containing cysteine-rich amino propeptide is secreted into the extracellular matrix and functions to bind bone morphogenetic proteins.33
TABLE 4-2 COLLAGEN FIBRIL DIAMETERS IN CARTILAGE | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Minor Collagens
Type IX Collagen. Type IX collagen can represent up to 10% of the articular collagen in the immature animal and 1% to 5% in the adult.34 Type IX collagen is classified as a FACIT collagen (fibril associated collagen with an interrupted triple-helix) forming heterotypic fibrils with types II and XI collagen. It is a heterotrimer [α1(IX) α2(IX) α3(IX)] composed of three chains being products of three distinct genes.35 Type IX collagen is different from type II collagen in that it has three triple-helical collagenous domains (COL1, 2, and 3) and four noncollagenous domains (NC1, 2, 3, and 4). These noncollagenous regions are more susceptible to proteolysis than collagenous domains. Overall, type IX is shorter than the fibrillar interstitial collagens and is stabilized by interchain disulfide bonds. There are long and short forms depending on the presence or absence of a large globular domain (NC4) at the amino terminal of the α1chain. Most unusual for a collagen molecule, it has a CS chain, leading to its additional classification as a proteoglycan. The type IX molecule is shown in Figure 4-7. A schematic representation of the type II collagen fibril with type IX associated is shown in Figure 4-8.
While the exact function of type IX collagen is not known, it is thought to function to limit the fibril diameter of the fibrillar collagen and is attached to the type II collagen by covalent hydroxylysyl pyridinoline and lysyl pyridinoline crosslinks. The possession of a large globular N-terminal domain and a CS chain are characteristics that suggest a potential to interact with other matrix components. Transgenic mice have been produced with a defective col9A1 gene that expresses a truncated α1(IX) chain.36,37 These animals develop pathological changes in the articular condyle of the knee joint which resembled those seen in osteoarthritis. Furthermore, mice that were homozygous for the mutation were found to have a mild chondrodysplasia. Another transgenic mouse
has been generated that lacks the α1(IX) chain.38 Surprisingly, even the homozygous mutants appear normal at birth, but later they develop a severe degenerative joint disease with similarities to human osteoarthritis. These findings suggest that changes in or lack of type IX collagen chains do not directly affect the gross appearance of the cartilage or its overall development. However, they may affect the organization of cartilage components on a molecular level leading to conditions such as chondrodysplasia or osteoarthritis, perhaps due to a loss of cartilage integrity which only becomes apparent when the cartilage is exposed to conditions of loading after birth. In humans where the cartilage must be maintained for a longer time, small defects in this collagen could contribute to early osteoarthritis, although no direct evidence for type IX defects leading to OA have been reported.
has been generated that lacks the α1(IX) chain.38 Surprisingly, even the homozygous mutants appear normal at birth, but later they develop a severe degenerative joint disease with similarities to human osteoarthritis. These findings suggest that changes in or lack of type IX collagen chains do not directly affect the gross appearance of the cartilage or its overall development. However, they may affect the organization of cartilage components on a molecular level leading to conditions such as chondrodysplasia or osteoarthritis, perhaps due to a loss of cartilage integrity which only becomes apparent when the cartilage is exposed to conditions of loading after birth. In humans where the cartilage must be maintained for a longer time, small defects in this collagen could contribute to early osteoarthritis, although no direct evidence for type IX defects leading to OA have been reported.
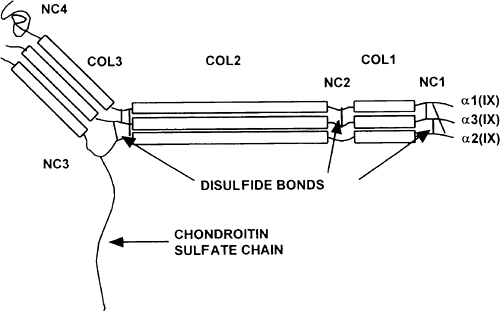 Figure 4-7 Diagram of a heterotypic fibril consisting of types II, IX and XI collagen. |
Collagen. Like type IX collagen, type XI is more abundant in immature cartilage from which it was originally isolated.40 It consists of three distinct polypeptide chains, α1(XI), α2(XI), and α3(XI), that form predominantly heterotrimeric molecules containing one of each chain. The α1(XI) and α2(XI) chains are products of separate genes, COL11A1 and COL11A2, respectively, and the proα3(XI) chain is a product of the COL2A1 gene, which codes for the α1(II) chain. The difference between the two chains is due to post-translational hydroxylation and glycosylation.41 Type XI is usually found in association with type II collagen in cartilaginous matrices (Fig. 4-7). In common with other fibrillar collagens, the α-chains of type XI collagen are synthesized as precursor procollagen chains consisting of a triple helical domain of more than 1,000 amino acids, with globular extensions at the amino and carboxy-termini. Type XI collagen has five major domains: NC1 (C-terminal, noncollagenous “hinge”-like region), NC2 (C-terminus), COL1 (major collagenous domain), COL2 (minor collagenous domain), and NC3 (N-terminal noncollagenous domain).
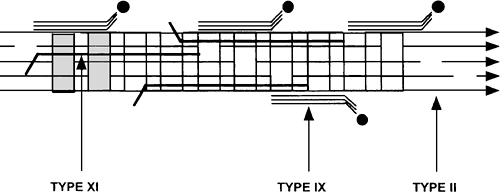 Figure 4-8 Diagram of a heterotopic fibril consisting of types II (COL2), IX (COL3) and XI (COL1) collagen. Boxes are triple helical domains and lines are globular protein domains. NC = noncollagenous domain; COL = Collagenous domain |
Processing of type XI collagen has been studied in embryonic chick sterna in vitro and is slower and more complex than that of collagen types I and II. All three chains of type XI collagen are initially processed at the C-terminal domains, which are linked through cysteines. α3(XI) undergoes one processing step at the N-terminal domain (removal of the signal peptide), which results in the fully processed matrix form of this chain.35 The other two chains undergo a two-step processing that result in portions remaining in the extracellular matrix. The matrix form of type XI is approximately 315 nm in length. The COL1 triple helical domain has binding sites which may be involved in interacting with other extracellular matrix components. α2(XI) and α2(XI) both have heparin binding sites and can also bind to aggrecan, the major cartilage proteoglycan, through these sites. It has been suggested that heparan sulfate-type XI collagen interactions may be of importance on the chondrocyte surface. Along the COL1 domain of α2(XI) there are three Arg-Gly-Asp (RGD) motifs which could contribute to chondrocyte surface interaction by binding to cellular integrins.
The NC3 noncollagenous globular domain of type XI collagen differs from those of collagen types I, II, and III in length and structure. The NC3 domains of proα1(XI) and proα2(XI) may be divided into two main subdomains. The N-terminal is a module rich in acidic residues, isolated originally from cartilage as a disulfide-bonded molecule called PARP (proline/arginine rich protein).42
C-terminal of PARP is a variable region (VR). Variations of this region occur as a result of alternative exon usage, conferring acidic or basic properties on the protein domain. The function of different domains is not presently known. The α3(XI) is the type IIB splice form of type II collagen.43,44 These N-terminal domains that are retained in the mature type XI molecule likely contribute to lateral aggregation of collagen molecules.
C-terminal of PARP is a variable region (VR). Variations of this region occur as a result of alternative exon usage, conferring acidic or basic properties on the protein domain. The function of different domains is not presently known. The α3(XI) is the type IIB splice form of type II collagen.43,44 These N-terminal domains that are retained in the mature type XI molecule likely contribute to lateral aggregation of collagen molecules.
The function of type XI collagen is thought to primarily lie in its role in fibril formation with type II collagen and interactions with components of cartilage proteoglycans and chondrocytes. Identification of mutations that cause specific chondrodysplasias have provided further evidence of the biological role of type XI collagen in cartilage and in skeletal morphogenesis. Mice homozygous for the auto-somal recessive chondrodysplasia (cho) mutation have abnormalities in cartilage, notably large collagen fibrils and loss of cohesion with increased ease of proteoglycan extraction. Both the COL11A1 and COL11A2 genes have also been implicated in forms of Stickler syndrome.45
Types III and V Collagens. Small amounts of type III collagen are seen in cartilage, primarily associated with type II collagen.46 In cell culture, human articular chondrocytes synthesize type III collagen.47 Type V collagen has been found in cartilage, particularly in older tissue48 where it replaces the α1(XI) collagen chain in the type XI molecule.
Type VI Collagen. Type VI collagen is not considered to be characteristic of cartilage, but is present in articular cartilage and located largely in the pericellular capsule around the chondrocytes. It may play a role in cell adhesion. Type VI collagen is not a fibrillar collagen, but contains a short triple-helical domain (105 nm) and N- and C-terminal globular domains that are very large and account for more than two thirds of the mass of the molecule. There are three α-chains (α1, α2, and α3), all with a multidomain structure: their globular domains contain modules homologous to von Willebrand factor A (vWFA). The type VI molecules assemble into dimers in an antiparallel fashion that aggregate laterally to form disulfide bonded tetramers. These associate noncovalently to form networks of microfibrils. The type VI microfibrils have a beaded appearance and are found in most connective tissues including articular cartilage where they are stabilized by interaction of the large globular domain of the α1(VI) chain with hyaluranon.49 In addition type VI collagen is rich in RGD sequences and has been shown to bind to the surface of many cells including chondrocytes.50 Type VI is distributed widely throughout the ECM in fetal cartilage but becomes restricted to the pericellular domain during growth.51 Type VI collagen is enriched in human OA cartilage and in cartilage from surgically induced OA in dogs.52 The role played by type VI collagen in normal tissue and in OA tissue is unknown.
Type X Collagen. Type X collagen is characteristic of hypertrophic chondrocytes in growth plate and is expressed primarily in this tissue with some localized in the calcified articular cartilage. Type X collagen is a homotrimer comprised of three identical α1(X) chains (Mr 59kDa) with 45 kDa of triple-helical collagenous domain flanked by two noncollagenous regions.53 The triple helix contains eight imperfections of the Gly-X-Y triplet structure allowing cleavage by MMP-1 at two sites.54 The accumulation of type X collagen mRNA and protein deposition in hypertrophic cartilage was found always to precede vascular invasion and mineralization of the matrix. It has generally been accepted that type X collagen is a product of chondrocytes undergoing hypertrophy, and collagen X expression is frequently used as a marker for chondrocyte hypertrophy. However, Ekanayake and Hall55 demonstrated that chondrocytes derived from chick mandibular ectomesenchyme, a normally permanent cartilage, can be induced to undergo maturation in culture, indicating that chondrocyte hypertrophy is not a prerequisite for collagen X expression. Also type X collagen has been immunolocalized in normal porcine, neonatal, and human articular cartilage, and in the mineralized fibrocartilage at the femoral insertion of the bovine medial collateral ligament where cell hypertrophy does not normally occur.56
Type X collagen may serve primarily as a structural element, either alone or in conjunction with other matrix components. The hypertrophic zone of the growth plate is structurally the weakest point within the growth plate by virtue of the increase in chondrocyte size and the decrease in the amount of type II collagen fibrils. It is generally accepted that collagen X provides a permissive matrix for chondrocyte hypertrophy, mineralization, and vascular invasion to occur during endochondral ossification. Type X collagen has been shown to have calcium binding properties57, to be associated with alkaline phosphatase58 and with matrix vesicles59 considered by some to be the initial site of mineral deposition in cartilage. However, it is safe to say that the exact role played by this collagen is not known.
Synthesis of type X collagen primarily by hypertrophic chondrocytes suggests that there must be a unique program of differentiation that is expressed only during the later stages of cartilage development. Due to the restricted expression of the gene, its regulation is of considerable interest. Lu Valle and associates have shown that the gene actually is driven by a very strong promoter and is regulated by multiple negative regulatory elements that aid in restricting expression in nonhypertrophic chondrocytes and other cells.60
Types XII and XIV Collagens. Types XII and XIV collagen have features in common with type IX collagen, but also have a very large noncollagenous amino-terminal domain.61 These collagens are closely associated with type I fibrillar collagen and are speculated to be located on the surface of type I collagen in a similar manner to the association of type IX with type II. The direct interaction of types XII and XIV and fibrillar type II has not been reported, although it is likely.
Other Molecules in Cartilage
Cartilage contains a large number of extracellular matrix proteins in addition to collagens and proteoglycans (Fig. 4-3). These have a wide range of roles, e.g., in facilitating matrix assembly, in maintaining the mechanical properties of the tissue, in sequestering growth factors and proteinases to specific compartments of the matrix, and in interacting with the cells important in the regulation of cellular activities. In several instances the functions of the proteins are
becoming known, while in many cases functional properties remain to be elucidated. The following description is focused on proteins where functional properties are becoming known.
becoming known, while in many cases functional properties remain to be elucidated. The following description is focused on proteins where functional properties are becoming known.
Thrombospondins (TSPs)
One of the major proteins in cartilage is cartilage oligomeric matrix protein (COMP), a member of the thrombospondin family as TSP-5.62,63,64 The thrombospondins are proteins built from a number of similar modules that share a number of functional properties. The proteins contain three (TSP 1 and 2) or five (TSP 3, 4, and 5) identical subunits. The subunits share a typical structure of their C-terminal part with a conserved globular domain, followed by a domain with seven calcium binding T3 repeats and a domain of three or four EGF-like T2 repeats. The 3D-structure of thrombospondin 1 has been resolved65 and shows that the T3 repeats contain the DxDxDGxxDxxD motif that binds two calcium ions. The repeats wrap around a core of central calcium ions. The T3 domain appears to form the C-terminal globular domain together with the very C-terminal cartilage oligomeric protein (CTD). The folding and structure of the chains of the thrombospondin proteins to a large extent depend on the large number of calcium ions bound with high affinity, particularly in the T3 domain. The total number of bound calcium ions is estimated to be 26 for the whole chain of thrombospondin 1.65
Another feature common to all the thrombospondins is the heptad repeat domain forming a coiled coil uniting the chains in the molecule.66 Disulfide bonds making the association covalent stabilize this structure. In COMP or thrombospondin 5, this domain is localized very close to the N-terminus of the protein. Different from COMP, the other thrombospondins contain a domain on the N-terminal side of the coiled coil linking the chains, with an overall folding similar to the G-domain of laminins.62 Thrombospondin 1 and 2 contain additional domains in the form of a von Willebrand factor C domain and a repeat T1 (properdin-like). These domains contribute cell binding to these proteins. Additional functions involve heparin binding which could also influence cell surface molecules in the form of syndecans. These domains also have roles in modulating cell behavior and particularly angiogenesis. One structure particularly relevant appears to be the heparin-binding motif present in the T1-domain. This may engage cell surface proteoglycans carrying heparan sulfate side chains.
The thrombospondins identified in cartilage are primarily TSP-1, TSP-3, and TSP-5/COMP, where COMP is predominant.67 The functions of this protein, most likely shared by the other two thrombospondins in the tissue, are being unraveled. The examples provided primarily make use of COMP as an example but are likely to apply to the other members of the TSP family, although in some cases when the multivalency of the protein is important there may be a difference between those with five and three chains, respectively.
The C-terminal globular domain mediates very tight KD = 10-9 binding to collagen type I or type II molecules with the ability to bind each of four sites with the same affinity.68 A role for this binding appears to be to accelerate and enhance collagen fibrillogenesis, where the COMP molecule appears to simultaneously bind several collagen molecules and arrange them in close proximity and facilitate their association upon forming fibrils.
COMP has been shown to also interact with collagen type IX, bound into collagen fibers such that particularly the col3 and NC4 domains protrude out from the fiber. In this case, the binding appears to be directed primarily to the noncollagenous domains, perhaps allowing the molecule to crossbridge neighboring fibers. The COMP also binds to fibronectin.69 It is not known whether this interaction occurs in vivo and will modulate functions such as interactions of the fibronectin at the cell surface.
The thrombospondins have been shown to bind cells via an RGD-motif present in the T3-domain.70 However, this motif appears to be primarily exposed in low calcium concentrations and a physiological role needs to be established. Indeed it appears that COMP from rat does not contain the RGD-sequence found in the human and bovine protein.63
A number of mutations, often point mutations, primarily in the T3 and some in the C-terminal domain of COMP have been shown to lead to pseudoachondroplasia and multiple epiphyseal dysplasia71 with growth disturbances. It appears that an important part of the defect is that the mutated molecule is retained in the endoplasmic reticulum (ER) of the chondrocytes thereby creating major deposits72 apparently affecting cell function possibly via dilation of the ER or by retaining other molecules via interactions. The total absence of the molecule in the extracellular matrix has apparently no or very limited effects on the tissue since the mouse where the COMP gene has been inactivated shows no detectable alterations in phenotype73 despite the absence of the protein.
COMP production and abundance in the matrix is markedly increased in early osteoarthritis, both at very early, preclinical stages and at late stages.67 The distribution of the protein is shifted from the interterritorial matrix of the normal adult cartilage to a pericellular environment, implying concomitant degradation and new deposition in a different compartment.
A novel observation that adds complexity to the understanding of thrombospondin functions is the finding in tendon samples of hetero-oligomers where a molecule contains subunits of both TSP-4 and TSP-5.74 This may not represent a form present in cartilage in view of the apparent lack of the protein in this tissue.
Matrilins
A family of proteins present in various tissues is the four matrilins, 1 to 4,75 which represent different gene products. They have in common that they are multimeric proteins, with three of four chains held together by a coiled coil domain. They contain four vWFA that are often seen in proteins involved in interactions. In cartilage, matrilins 1 and 3 are primarily found.76 They contain two and one such domains per chain, respectively. Interestingly, there are also matrilin molecules that contain a mixture of chains of type 1 and 3.77
The vWFA domain contributes binding properties to the matrilins, involving the fibrillar collagens as well as a number of other molecules.78,79 In proof of the relevance of
some of these interactions, it has been possible to use tissue homogenization of cartilaginous tissues such as the Swarm rat chondrosarcoma to isolate complexes of collagen type VI with other sets of molecules still attached. Thus biglycan or decorin were bound at the C-terminal globular domains of the collagen and were in turn binding a molecule of the matrilin family, where matrilins-1, -2, and -3 were identified, binding with one of the subunits.80 The matrilin in turn binds other molecules such as a collagen fiber or the proteoglycan aggrecan.80,81 It thus appears that the combination of a small proteoglycan in the form of decorin or biglycan and a matrilin can act as a linker molecule between the collagen VI beaded filaments to other major structural assemblies in the tissue. The finding of bound procollagen type II at the linker module80 may indicate that there is also a function in regulating assembly of the collagen fibers.
some of these interactions, it has been possible to use tissue homogenization of cartilaginous tissues such as the Swarm rat chondrosarcoma to isolate complexes of collagen type VI with other sets of molecules still attached. Thus biglycan or decorin were bound at the C-terminal globular domains of the collagen and were in turn binding a molecule of the matrilin family, where matrilins-1, -2, and -3 were identified, binding with one of the subunits.80 The matrilin in turn binds other molecules such as a collagen fiber or the proteoglycan aggrecan.80,81 It thus appears that the combination of a small proteoglycan in the form of decorin or biglycan and a matrilin can act as a linker molecule between the collagen VI beaded filaments to other major structural assemblies in the tissue. The finding of bound procollagen type II at the linker module80 may indicate that there is also a function in regulating assembly of the collagen fibers.
The matrilins also appear to be able to associate into networks on their own,82 probably involving their vWFA domains. The exact relevance of these interactions to tissue function is not known. The genes for the various matrilins have been inactivated, without providing a major phenotype.83,84,85 However, patients with mutations of the matrilins show multiple epiphyseal dysplasia (MED) with skeletal growth abnormalities,86 perhaps via mechanisms similar to those of COMP mutations.
Matrilin 3 is present in most cartilages, while matrilin-1 shows a very restricted distribution. Thus the protein is present in young growing tissue and also very abundant in cartilage like that in the trachea. At the same time the protein is not detected in articular cartilage, not even at a level of 1000-fold lower, neither in the intervertebral disc structures.87 When mice or rats with certain genetic backgrounds are immunized with the protein, they develop severe respiratory failure due to autoimmune reaction and inflammation in the trachea.88 The animals also have engagement of other tissues where the protein is found, such as the nasal cartilage. However, they show no joint disease.
CILP
CILP is the acronym for cartilage intermediate layer protein, which was first identified as a component in articular cartilage that is upregulated in early osteoarthritis.67 The protein was found to be primarily present in the interterritorial matrix in the middle of articular cartilage from adult individuals.89 The cloned and sequenced protein was shown to represent a larger molecule.90 This precursor form was cleaved to two parts upon secretion from the cells. The N-terminal portion represents the previously isolated CILP protein with a molecular mass of 78.5 kDa, while the C-terminal part shows a very different structure, homologous to the NTPPHase enzyme, molecular mass 51.8 kDa, not including posttranslational modifications. Whether this putative enzyme actually has activity is not clear, although found data indicate that it is not active.91 There is a related protein referred to as CILP-2, initially as a genomic sequence. It is homologous to CILP (now CILP-1) and shares about 50% of the structure. CILP-2 has an N-terminal part related to CILP and a C-terminal part related in sequence to NTPPHase. To avoid confusion, the N-terminal parts are now referred to as CILP 1-1 and CILP 2-1 while the C-terminal parts are CILP 1-2 and CILP 2-2. To date we know very little about the role of CILP in cartilage. It has, however, been shown that it increases in OA and may serve as an antigen in joint disease.92 It has been suggested that CILP may have a role in regulating the activity of insulin-like growth factor on cells.91
Leucine-Rich Repeat Proteins
The leucine-rich repeat (LRR) proteins in the extracellular matrix can be divided into families with sets of members that differ in gene organization and structural features. They share a common feature with a central domain of 10 or 11 (in decorin, biglycan, and asporin 12) repeats, each with some 25 amino acids and with leucine residues at conserved locations.93,94,95 This central repeat region is surrounded by disulfide loop structures, where there are two disulfide bridges on the N-terminal side and one on the C-terminal side. Three of the LRR proteins regularly possess one or two glycosaminoglycan (GAG) chains and are therefore proteoglycans. Many of the others contain one or several N-glycosidically linked KS chains, but this is a variable between species, tissues, and age of the animal. Those with GAG chains have been termed small leucine-rich repeat proteoglycans (SLRPs).
It is likely that the 3D structure of this leucine rich domain shares the features of the one determined for decorin96 and verified for biglycan.97 The curved core of the repeat region exposes a parallel β-sheet surface on the concave side. This face is involved in interactions between two molecules that results in the formation of dimers where the N-terminal end of one molecule is located close to the middle of the concave face of the other, thus exhibiting a very large interacting surface. If all the LRR molecules are involved in such a dimer formation, this will influence how these proteins interact with other molecules. The LRR portion of all the proteins appears to bind with high affinity to fiber-forming collagens (DEC, BIGN, FM, LUMICAN, PRELP, CHAD), although for most of the proteins, the exact binding sites have not been identified. The sites along the collagen molecule appear from preliminary data to be different for many of the proteins, with the possible exception of fibromodulin and lumican.98 In several cases, it appears that there is more than one site along the collagen (PRELP, CHAD).99,100 It also appears that other interactions involve the LRR-repeat domain, including binding of growth factors within the TGF- β family.101,102 It also appears that interactions with, for example, the matrilins and collagen type VI described involve the LRR-region,80,103 but the exact specificity is not known. The major differences between the molecules are found in the N-terminal and C-terminal portions outside the disulfide loops flanking the LRR-repeats. There are some characteristic differences between each family of molecules, described below. At the same time, there are distinct differences between individuals in a given family.
Four distinct classes of SLRPs have been described (Fig. 4-9) (LLR), based upon general structural103 class I SLRPs decorin93 and biglycan,104 and LRR protein Asporin95
show high structural similarity. Class II includes fibromodulin,105 lumican,106 keratocan,107 PRELP,108 and osteoadherin.109 Class II can be subdivided into subclasses, with fibromodulin and lumican showing greater relatedness, as well as keratocan and PRELP. Osteoadherin may represent a member of a third subclass within Class II.109 Class III contains epiphican110,111 (also known as PG-Lb) and osteoglycin.112 PRELP should most likely be considered an LRR glycoprotein as little evidence exists that it is substituted with GAG chains. Another LRR protein, chondroadherin, particularly prominent in cartilage, has a distinct gene arrangement distinguishing this protein to a separate class. It contains neither GAG chains nor N-glycosidically linked oligosaccharides.
show high structural similarity. Class II includes fibromodulin,105 lumican,106 keratocan,107 PRELP,108 and osteoadherin.109 Class II can be subdivided into subclasses, with fibromodulin and lumican showing greater relatedness, as well as keratocan and PRELP. Osteoadherin may represent a member of a third subclass within Class II.109 Class III contains epiphican110,111 (also known as PG-Lb) and osteoglycin.112 PRELP should most likely be considered an LRR glycoprotein as little evidence exists that it is substituted with GAG chains. Another LRR protein, chondroadherin, particularly prominent in cartilage, has a distinct gene arrangement distinguishing this protein to a separate class. It contains neither GAG chains nor N-glycosidically linked oligosaccharides.
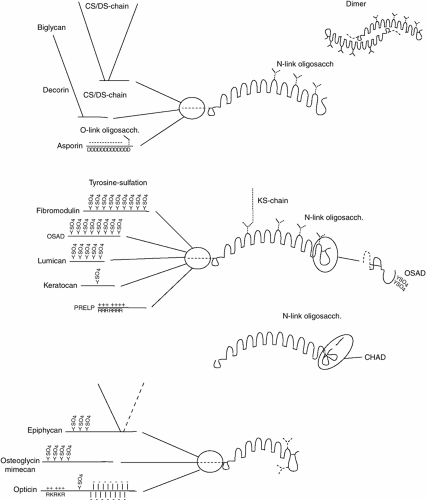 Figure 4-9 Schematic depiction of the four groups of LRR proteins found in the extracellular matrix. The curved shape of the core protein and dimer formation observed by x-ray crystallography for biglycan and decorin are indicated. The variable N-terminal region (to the left) of the molecules in each group are indicated separately with glycosaminoglycan chains as filaments (black), tyrosine sulfate as a SO4, and N-linked oligosaccharides as a Y-structure (black). In some cases the N-linked oligosaccharide may exist in the form of a KS chain, with a longer arm representing the disaccharide repeat. Clusters of basic (e.g., PRELP) amino acids are denoted by R and a plus (+) sign, while arrays of acidic amino acids (in asporin) are denoted by a D and a minus (-) sign. The mucin like cluster of O-linked oligosaccharides in opticin are denoted as fat rods with a minus (-) sign. In the cases of osteoadherin (OSAD) and chondroadherin (CHAD), the C-terminal part is different as is indicated. |
Decorin, Biglycan, and Asporin. The members of the first family are decorin, biglycan, and asporin. Decorin has a single N-terminal site for addition of a CS or DS GAG chain, and three potential sites for N-linked oligosaccharide substitution.114 Decorin (as well as biglycan and asporin) is synthesized with amino terminal propeptides that apparently may be removed intracellularly during biosynthesis114 or extracellularly115 (as both processed and proforms can be detected in culture medium and cartilage extracellular matrix.115 A role for decorin in collagen fibrillogenesis is supported by the observation that decorin-null animals demonstrate skin fragility.115a The reduction in tensile strength could be associated with abnormal collagen fiber formation in the skin. Decorin has also been shown to have a high affinity for TGF-β, which may permit decorin to sequester these growth factors in the extracellular matrix.101 Independent of the interaction of decorin with TGF-β, decorin may function to inhibit cellular proliferation by acting directly upon a signal-transduction pathway leading to activation of cyclin-dependent kinase inhibitors.116a
Biglycan has two N-terminal sites for CS or DS GAG substitution, and two potential N-linked oligosaccharide sites. Biglycan, unlike decorin, has been shown to be present in cartilage as intact and N-terminally degraded forms with increasing age.116b Biglycan-deficient mice117 show diminished bone mass with increasing age, and are deficient in their capacity to form bone. This is likely due to a diminished capacity to produce marrow stromal cells (bone cell precursors). These cells also have reduced response to TGF-β, reduced collagen synthesis, and increased apoptosis.
Asporin differs from decorin and biglycan in that it contains an array of aspartates in its N-terminal that show a polymorphism with a variable number from 11 up to 15 such residues.96 Studies of individuals with osteoarthritis indicate that in some populations there is a correlation of a higher risk for the disease with 13 aspartates118 while in other cohorts this relation has not been verified.101,119,120 Like other LRR-proteins, asporin is expected to bind to fibril-forming collagens type I and II. This has not been verified although some preliminary data indicate that this is true. It has been suggested that asporin can bind TGF-β118 similarly to decorin, biglycan, and fibromodulin.101 Such an interaction could contribute to sequestering the growth factor to certain structures in the matrix to be released, for example, upon degradation of these structures in pathology. The released factor can then activate the cells to an adequate repair response.
Fibromodulin, Lumican, Keratocan, Osteoadherin, and PRELP. Most members of this family of proteins contain tyrosine sulfate residues in the N-terminal extension. Fibromodulin is the most prominent example with up to nine such tyrosine sulfates over a short domain121 providing the molecule with an extremely anionic domain. Because fibromodulin is present bound at the collagen fiber surface in the tissue,122 it is likely that this N-terminal extension is available for interactions with more cationic motifs on surrounding structures. Indeed the availability of this domain is indicated by the fact that upon stimulation of degradation processes in cartilage by IL-1, a specific cleavage accomplished by MMP-13 will release almost the entire tyrosine sulfate domain123 and leave the rest of the protein bound to collagen. The large variability in the number of tyrosine sulfate residues within a given preparation provides for variable potential functions. Fibromodulin contains additional anionic structures in the form of KS chains,124,125 but this is not obligatory and varies between species and age of the individual.
Osteoadherin109,126 contains up to eight tyrosine sulfates that show a different clustering compared to fibromodulin.121 Much less is known about this protein that is restricted to bone but also made by hypertrophic chondrocytes. It does bind cells via their αv β3 integrins,126 but the details of this interaction are not known. Osteoadherin differs from the other members of this family by having an extensive C-terminal portion containing two tyrosine sulfates.121 Also osteoadherin contains KS chains depending on tissue.126 Lumican127 contains a maximum of four tyrosine sulfates in the N-terminus,121 and contains KS in some tissues it appears to bind to the same site on collagen fibers as fibromodulin although with somewhat lower affinity.98
In support of the collagen binding of these molecules, the fibromodulin- and lumican-null mice show distinct alterations of the collagen fiber assembly.128,129,130
Keratocan107 has only one potential site for tyrosine sulfation, but shares many other features with the other molecules in the family.
PRELP131 is distinct in containing an N-terminal extension with clusters of basic amino acids forming a domain that binds heparin with high affinity. Its affinity is highest for heparin with three O-sulfates per disaccharide. This protein was originally found in cartilage,132 but later studies have revealed its presence at basement membranes where it appears to bind to the heparan sulfate chains of, for example, perlecan via the specific N-terminal binding domain. At the same time the protein can bind to collagen serving to provide anchorage of basement membranes to underlying tissues. The protein is relatively abundant in cartilage, but its exact role is not known. Cartilage indeed contains perlecan as one potential interaction partner133 and a role in network formation by simultaneous binding to the collagen fibers is likely.
Chondroadherin. Chondroadherin113 forms its own family and differs from the other LRR proteins by having a double disulfide loop on the C-terminal side of the repeat domain. Another difference is that this protein has
no N-terminal extension, while a short basic extension in the C-terminal should contribute functional properties, possibly involving interactions with anionic constituents like the GAG chains. The protein is also unique among the LRR-proteins in having no post-translational modifications.134 The protein has been shown to bind to the integrin α2 β1, but not to any of the other collagen binding integrins.135 Upon binding, the cells do not spread, but signals are induced, e.g., ERK-phosphorylation. Interestingly, the protein can also bind to fibril-forming collagen types I and II.100 Indeed, complexes with chondroadherin bound at two sites along the collagen II molecule could be isolated from cartilage after induction of endogenous proteinases. It is possible that chondroadherin can provide a bridging from the cell to collagen in the surrounding matrix. At this time little is know of the exact function of the protein in cartilage and the role of the interactions with the integrin has not been elucidated. Chondroadherin is quite restricted to cartilage. The protein is found particularly enriched in the growth plate and particularly in a zone corresponding to the prehypertrophic area.136 Novel findings have identified expression of the protein in several structures of the eye and in other tissues including some neurological.137
no N-terminal extension, while a short basic extension in the C-terminal should contribute functional properties, possibly involving interactions with anionic constituents like the GAG chains. The protein is also unique among the LRR-proteins in having no post-translational modifications.134 The protein has been shown to bind to the integrin α2 β1, but not to any of the other collagen binding integrins.135 Upon binding, the cells do not spread, but signals are induced, e.g., ERK-phosphorylation. Interestingly, the protein can also bind to fibril-forming collagen types I and II.100 Indeed, complexes with chondroadherin bound at two sites along the collagen II molecule could be isolated from cartilage after induction of endogenous proteinases. It is possible that chondroadherin can provide a bridging from the cell to collagen in the surrounding matrix. At this time little is know of the exact function of the protein in cartilage and the role of the interactions with the integrin has not been elucidated. Chondroadherin is quite restricted to cartilage. The protein is found particularly enriched in the growth plate and particularly in a zone corresponding to the prehypertrophic area.136 Novel findings have identified expression of the protein in several structures of the eye and in other tissues including some neurological.137
Proteins with a Shorter LRR-repeat region (opticin, mimecan/osteoglycin, and epiphycan). There is also a family of proteins/proteoglycans with only six or seven LRR repeats. This family consists of epiphycan, mimecan, and opticin. Available data indicate that these proteins also bind to fibril-forming collagens.138 This seems to be a feature of the LRR proteins. Also these proteins have N-terminal extensions with different properties. Opticin139,140 originally isolated from the vitreous of the eye is present in cartilage and is quite unique in that it contains a mucin stretch with O-glycosidically linked oligosaccharides. Further to the N-terminus, the protein contains a stretch rich in basic amino acids and apparently having the ability to bind to heparin and other GAG.141 This molecule may add to the network and quality of the cartilage matrix.
Mimecan142 contains an N-terminal domain with putative substitution with tyrosine sulfate. In addition to this anionic domain, the molecule when present in cornea contains KS. A smaller version of mimecan, osteoglycin, lacks the tyrosine sulfate domain. Epiphycan111 contains both a short stretch of tyrosine sulfate and up to two CS/dermatan sulfate chains in its N-terminal extension.
Tenascin-C
Tenascin-C is a member of the tenascin family with four members (C, R, W, X)143 having different tissue distributions. Tenascin-C is the one of these molecules with a significant presence in cartilage, particularly during growth and in pathology.144,145 The protein is also produced by fibroblasts including those in tumor stroma, smooth muscle cells, and in the nervous system.143 Tenascin-C forms a homo-hexamer, originally referred to as hexabrachion, where the chains are linked via a heptad repeat close to the N-terminus. Tenascin-C is present in several differently spliced forms, where the overall structure is similar. The C-terminal end of the chain contains a fibrinogen globe followed by a variable number (at least 8) of fibronectin type III repeats and some 15 EGF-like repeats.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


