Cardiomyopathy
Jeffrey A. Towbin
Neil E. Bowles
Cardiomyopathies in children are major causes of morbidity and mortality, and since the late 1980s, limited improvements in outcome have been reported. However, on the basis of improvements in the understanding of several of the major forms of cardiomyopathy in adults, as well as the treatment of these disorders, numerous concepts have been tested (or are currently being tested) in childhood forms of these disorders. In addition, various forms of cardiomyopathy commonly present early in
childhood, and these disorders are being described in detail by pediatric cardiologists.
childhood, and these disorders are being described in detail by pediatric cardiologists.
The major classifications of the forms of cardiomyopathies include (a) dilated cardiomyopathy (DCM), which includes approximately 55% of all cardiomyopathies; (b) hypertrophic cardiomyopathy (HCM), which accounts for approximately 35% of all cardiomyopathies; (c) restrictive cardiomyopathy (RCM), an uncommon form accounting for fewer than 5% of cases; and (d) arrhythmogenic right ventricular cardiomyopathy (ARVC), an uncommon form of disease as well, particularly in children. An unclassified form of cardiomyopathy, left ventricular noncompaction (LVNC), is considered to be quite rare, but recent evidence suggests that this disorder actually is relatively common but only rarely diagnosed.
These classified and unclassified disorders generally are considered diseases affecting primarily systolic function (DCM affecting the left ventricle, ARVC affecting the right ventricle), diastolic function (HCM and RCM), or both (LVNC). Based on these functional considerations, therapies have been devised that focus on affecting systole or diastole directly or indirectly.
Since the late 1980s, advances made in molecular genetics and mouse modeling efforts have resulted in modifications of our understanding of these disorders, as well as in therapies designed to treat these disorders. In this chapter, the clinical description, therapies, molecular basis, and functional abnormalities of these disorders are discussed. The goal of this chapter is to acquaint the reader with these disorders and a variety of new considerations regarding form and function of the heart and the potential new diagnostic and therapeutic horizons of the future for these potentially devastating diseases.
EPIDEMIOLOGY
The cardiomyopathies are heart muscle disorders associated with significant rates of morbidity and mortality. These disorders are classified by the World Health Organization (WHO) into four forms: (a) DCM, (b) HCM, (c) RCM, and (d) ARVC. More recently, another cardiomyopathy, LVNC, has gained attention, although it does not meet criteria for a separate classification currently.
The most common cardiomyopathy is DCM, accounting for approximately 55% of cases. HCM is second most common, accounting for approximately 35%, with the remaining forms accounting for approximately 5% or less in each case. The importance of these disorders lies in that they are responsible for a high proportion of cases of congestive heart failure (CHF) and sudden death, as well as the need for cardiac transplantation. The mortality rate in the United States from cardiomyopathy is greater than 10,000 deaths/year, with DCM being the major contributor to this death rate. The total cost of health care in the United States focused on cardiomyopathies is in the billions of dollars, and only limited success has been achieved. To achieve improved care and outcomes in children and adults, understanding of the causes of these disorders has been sought in earnest during the past decade.
PATHOGENESIS AND PATHOPHYSIOLOGY
HCM and RCM are primarily diastolic disorders, whereas DCM and ARVC are primarily systolic disorders. LVNC is a combination overlap disorder with evidence of both systolic and diastolic dysfunction. The molecular basis of HCM has been well studied since the early 1990s, and to date, 11 genes have been identified as being causes of disease. Nearly all these genes encode proteins comprising the sarcomere, including beta-myosin heavy chain, alpha-tropomyosin, cardiac troponin T, cardiac troponin I, myosin-binding protein C, myosin essential light chain, myosin regulatory light chain, actin, muscle LIM protein, and titin. Hence, the disease now is considered a disease of the sarcomere. One recently identified gene appears to question whether the sarcomere is the only region of the heart causing HCM when altered. This gene, called adenosine monophosphate (AMP)-kinase (AMPK), is important in metabolic regulation, participating in energy utilization, and its function appears to be similar to that seen in a glycogen storage disease.
TABLE 283.1. DILATED CARDIOMYOPATHY GENETICS | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
DCM has become a popular target of research during the past 7 to 8 years, with multiple genes identified during that period (Table 283.1). These genes appear to encode two major subgroups of proteins, cytoskeletal and sarcomeric. The cytoskeletal proteins identified to date include dystrophin, desmin, lamin A/C, delta-sarcoglycan, metavinculin, muscle LIM protein, and alpha-actinin. In the case of sarcomere-encoding genes, the same genes identified for HCM appear to be implicated in select patients. A new gene, cypher/ZASP, a Z-line protein, has been identified recently as well. In addition, two genes with uncertain mechanisms (including phospholamban and G4.5/Tafazzin) also are reported. Another form of DCM, the acquired disorder viral myocarditis, has the same clinical features as DCM, including heart failure, arrhythmias, and conduction block. The most common causes of myocarditis are viral and include the enteroviruses (coxsackieviruses and echovirus), adenoviruses, parvovirus B19, and other cardiotropic viruses. Evidence suggests that viral myocarditis and DCM (genetic) have similar mechanisms of disease based on the proteins targeted.
RCM, ARVC, and LVNC recently were recognized scientifically, but progress is being made in unraveling the underlying mechanisms of these disorders. Once the cardiomyopathies are understood at the molecular and cellular levels, targeted therapies can be developed in addition to current strategies, with the hope for improved outcomes. In this chapter, we review the clinical and scientific characteristics of these disorders and describe potential novel therapeutic approaches for future consideration. To understand the mechanisms responsible for the development of the clinical phenotype, an understanding of normal cardiac anatomy is necessary (Box 283.1 and Fig. 283.1). In addition, it is necessary to understand how other factors contribute to cardiomyopathy.
BOX 283.1. Normal Cardiac Structure
Cardiac muscle fibers are composed of separate cellular units (myocytes) connected in series. In contrast to skeletal muscle fibers, cardiac fibers do not assemble in parallel arrays but bifurcate and recombine to form a complex three-dimensional network. Cardiac myocytes are joined at each end to adjacent myocytes at the intercalated disc, the specialized area of interdigitating cell membrane (see Fig. 283.1). The intercalated disc contains gap junctions (containing connexins) and mechanical junctions, composed of adherens junctions (containing N-cadherin, catenins, and vinculin) and desmosomes (containing desmin, desmoplakin, desmocollin, and desmoglein). Cardiac myocytes are surrounded by a thin membrane (sarcolemma), and the interior of each myocyte contains bundles of myofibrils arranged longitudinally. The myofibrils are formed by repeating sarcomeres, the basic contractile units of cardiac muscle composed of interdigitating thin (actin) and thick (myosin) filaments (see Fig. 283.1) that give the muscle its characteristic striated appearance. The thick filaments are composed primarily of myosin but additionally contain myosin binding proteins C, H, and X. The thin filaments are composed of cardiac actin, alpha-tropomyosin (alpha-TM), and troponins T, I, and C (cTnT, cTnI, cTnC). In addition, myofibrils contain a third filament formed by the giant filamentous protein, titin, which extends from the Z-disc to the M-line and acts as a molecular template for the layout of the sarcomere. The Z-disc at the borders of the sarcomere is formed by a lattice of interdigitating proteins that maintain myofilament organization by cross-linking antiparallel titin and thin filaments from adjacent sarcomeres (see Fig. 283.1). Other proteins in the Z-disc include alpha-actinin, nebulette, telethonin/T-cap, capZ, microsomal lipoprotein (MLP), myopalladin, myotilin, Cypher/ZASP, filamin, and FATZ.
Finally, the extrasarcomeric cytoskeleton, a complex network of proteins linking the sarcomere with the sarcolemma and the extracellular matrix (ECM), provides structural support for subcellular structures and transmits mechanical and chemical signals within and between cells. The extrasarcomeric cytoskeleton has intermyofibrillar and subsarcolemmal components, with the intermyofibrillar cytoskeleton being composed of intermediate filaments, microfilaments, and microtubules. Desmin intermediate filaments form a three-dimensional scaffold throughout the extrasarcomeric cytoskeleton, with desmin filaments surrounding the Z-disc, allowing for longitudinal connections to adjacent Z-discs and lateral connections to subsarcolemmal costameres. Microfilaments composed of nonsarcomeric actin (mainly gamma-actin) also form complex networks linking the sarcomere (via alpha-actinin) to various components of the costameres. Costameres are subsarcolemmal domains located in a periodic, grid-like pattern, flanking the Z-discs and overlying the I-bands, along the cytoplasmic side of the sarcolemma. These costameres are sites of interconnection between various cytoskeletal networks linking sarcomere and sarcolemma and are thought to function as anchor sites for stabilization of the sarcolemma and for integration of pathways involved in mechanical force transduction. Costameres contain three principal components: the focal adhesion-type complex, the spectrin-based complex, and the dystrophin/dystrophin-associated protein complex (DAPC). The focal adhesion-type complex, composed of cytoplasmic proteins (i.e., vinculin, talin, tensin, paxillin, zyxin), connects with cytoskeletal actin filaments and with the transmembrane proteins alpha-, beta-dystroglycan, alpha-, beta-, gamma-, delta-sarcoglycans, dystrobrevin, and syntrophin. Several actin-associated proteins are located at sites of attachment of cytoskeletal actin filaments with costameric complexes, including alpha-actinin and the muscle LIM protein, MLP. The C-terminus of dystrophin binds beta-dystroglycan (see Fig. 283.1), which, in turn, interacts with alpha-dystroglycan to link to the ECM (via alpha-2-laminin). The N-terminus of dystrophin interacts with actin. Also notable, voltage-gated sodium channels colocalize with dystrophin, beta-spectrin, ankyrin, and syntrophins, and potassium channels interact with the sarcomeric Z-disc and intercalated discs. Because arrhythmias and conduction system diseases are common occurrences in children and adults with dilated cardiomyopathy, this factor could play an important role. Hence, disruption of the links from the sarcolemma to ECM at the dystrophin C-terminus and those to the sarcomere and nucleus via N-terminal dystrophin interactions could lead to a domino-effect disruption of systolic function and development of arrhythmias.
Role of Cytokines in Myocarditis and Dilated Cardiomyopathy
During the last few years, considerable interest has been shown in the role of cytokines in the pathogenesis of myocarditis and DCM. Animal studies have suggested that a relationship may exist between subclinical viral infection and later development of DCM. This process is presumed to occur by an autoimmune-like mechanism triggered by the initial viral insult. Several murine models have been studied that suggest that cytokine-mediated modulation of the immune response to viral infection may lead to induction of chronic autoimmune myocarditis.
Among their many immunomodulatory activities, cytokines contribute to regulation of antibody production and maintenance of “self-tolerance.” Certain susceptible murine strains, when infected with coxsackievirus B3, are known to develop myocyte necrosis and an acute inflammatory response consisting mainly of neutrophils and macrophages within the heart. After the initial viral infection, resolution of inflammation eventually occurs. In other strains, however, a second autoimmune phase of myocarditis appears later with findings of diffuse mononuclear cell infiltrates within the heart. These mononuclear cells are a significant source of the cytokines interleukin-1 (IL-1) and tumor necrosis factor-alpha (TNF-alpha), and work by Henke and associates demonstrated that release of large amounts of TNF-alpha and IL-1beta by human monocytes exposed to coxsackievirus B3 occurs. Both these cytokines are known to participate in leukocyte activation, which may be beneficial in promoting a specific lymphocyte response to viral infection. However, these cytokines also may promote cardiac fibroblast activity, and, therefore, researchers have speculated that local secretion of cytokines in the myocardium perpetuates the inflammatory process, which secondarily leads to the fibrosis associated with DCM and resultant deterioration of cardiac function. Studies by Gulick and associates
initially implicated IL-1 and TNF-alpha as potential inhibitors of cardiac myocyte beta-adrenergic responsiveness, and further studies showed IL-1 and TNF-alpha to be the macrophage factors mediating this effect. In particular, TNF-alpha has been studied in some detail, resulting in reports of elevated TNF-alpha levels in the serum of patients with chronic heart disease, including a subset of patients with myocarditis or DCM. TNF-alpha is able to potentiate the immune response and induce apoptosis in cells, both of which appear to hold special importance in the pathogenesis of myocarditis. Other inflammatory mediators, including IL-1 and granulocyte colony-stimulating factor, also are elevated in patients with myocarditis and have received attention as well. Other studies have suggested that inflammatory cytokines actually may cause a direct negative inotropic response.
initially implicated IL-1 and TNF-alpha as potential inhibitors of cardiac myocyte beta-adrenergic responsiveness, and further studies showed IL-1 and TNF-alpha to be the macrophage factors mediating this effect. In particular, TNF-alpha has been studied in some detail, resulting in reports of elevated TNF-alpha levels in the serum of patients with chronic heart disease, including a subset of patients with myocarditis or DCM. TNF-alpha is able to potentiate the immune response and induce apoptosis in cells, both of which appear to hold special importance in the pathogenesis of myocarditis. Other inflammatory mediators, including IL-1 and granulocyte colony-stimulating factor, also are elevated in patients with myocarditis and have received attention as well. Other studies have suggested that inflammatory cytokines actually may cause a direct negative inotropic response.
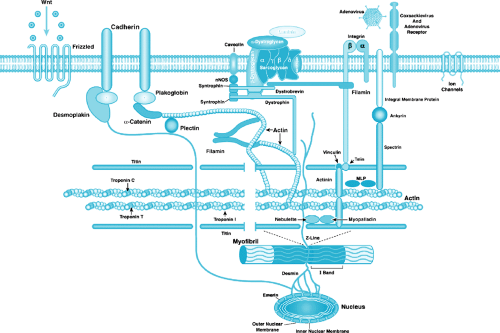 FIGURE 283.1. Cardiac myocyte cytoarchitecture. Schematic of the interactions between dystrophin and the dystrophin-associated proteins in the sarcolemma and intracellular cytoplasm (dystroglycans, sarcoglycans, syntrophins, dystrobrevin, sarcospan) at the C-terminal end of the dystrophin. The integral membrane proteins interact with the extracellular matrix via alpha-dystroglycan-laminin alpha-2 connections. The N-terminus of dystrophin binds actin and connects dystrophin with the sarcomere intracellularly, the sarcolemma, and the extracellular matrix. N, amino terminus; C, carboxy terminus; MLP, muscle LIM protein. |
Role of Cell Adhesion Molecules in Myocarditis and Dilated Cardiomyopathy
Molecules now known to play major roles in many processes of inflammation, the distinct classes of cell adhesion molecules (CAMs), also may play roles in the pathogenesis of myocarditis. One molecule that is well known to play a major role in cell-cell adhesion, particularly leukocyte adherence and transendothelial migration, is intercellular adhesion molecule-1 (ICAM-1). ICAM-1 is a member of the immunoglobulin supergene family of CAMs and is a single-chain glycoprotein of 80 to 115 kDa with an extracellular domain composed of five immunoglobulin-like repeats. ICAM-1 is expressed predominantly on endothelial cells but also on fibroblasts, epithelial cells, mucosal cells, lymphocytes, monocytes, and cardiac myocytes after inflammatory injury. Expression of ICAM-1 on endothelial cells is known to be upregulated by cytokines such as IL-1 and TNF-alpha. A well-established binding ligand of ICAM-1 is lymphocyte function-associated antigen-1 (LFA-1), a molecule that is part of the beta-2 integrin family and consists of a 180-kDa alpha subunit (CD11a) and a 95-kDa beta subunit (CD18). LFA-1 is expressed on virtually all leukocytes, including monocytes. The adhesive interaction between LFA-1 and ICAM-1 is known to mediate adhesion-dependent helper T-cell, cytotoxic T-cell, and NK cell functions. Antibody to LFA-1 has been used for therapy in animal models of myocarditis, with resultant blockade of the inflammatory response.
Apoptosis
Apoptosis, or programmed cell death, has an important role in embryogenesis, tissue homeostasis, and regulation of immunologic responses, among normal physiologic processes, and it is associated with the growth and regression of tumors. Cells undergoing apoptosis exhibit characteristic morphologic and biochemical features, including chromatin aggregation, nuclear and cytoplasmic aggregation, and formation of apoptotic
bodies resulting from the partition of the cytoplasm and nucleus into membrane bound-vesicles. These apoptotic bodies are rapidly phagocytosed by adjacent macrophages or epithelial cells, without resulting in an inflammatory response. Apoptotic cells are detectable by terminal transferase labeling (terminal deoxynucleotide transferase-mediated biotin-deoxyuridine triphosphate nick end labeling [TUNEL]) in myocardial tissue samples from patients with DCM. It has been shown that as many as 0.1% of cells stained positive by this technique.
bodies resulting from the partition of the cytoplasm and nucleus into membrane bound-vesicles. These apoptotic bodies are rapidly phagocytosed by adjacent macrophages or epithelial cells, without resulting in an inflammatory response. Apoptotic cells are detectable by terminal transferase labeling (terminal deoxynucleotide transferase-mediated biotin-deoxyuridine triphosphate nick end labeling [TUNEL]) in myocardial tissue samples from patients with DCM. It has been shown that as many as 0.1% of cells stained positive by this technique.
Numerous viruses have been implicated in the induction of apoptosis, including human immunodeficiency virus (HIV), Epstein-Barr virus (EBV), and adenovirus. Apoptotic cells have been detected in myocardial sections from patients with adenovirus-associated myocarditis and DCM, the areas of staining are usually focal and a number of positive-staining areas may be detected within each section. Within such areas, as many as 1% of cells may stain positive, including myocytes, infiltrating inflammatory cells, and endothelial cells. In the tissue sections from control patients, either unstained or sporadic (one or two per section) stained cells may be detected. These data suggest a relationship between infection of the myocardium by adenovirus and the onset of apoptosis, which could result in pathologic processes associated with myocarditis and DCM. Further, numerous inflammatory cells may be seen to be undergoing apoptosis. Although this finding could reflect the natural defense mechanism of the host against the virus, it also raises the possibility of virus-induced apoptosis as a mechanism of immune system avoidance. Strand and associates reported that in tumors, infiltrating immune cells are destroyed by the induction of apoptosis through the expression of Fas ligand on the tumor cell that binds Fas on the lymphocyte.
DISORDERS OF VENTRICULAR SYSTOLIC DYSFUNCTION
Dilated Cardiomyopathy
Epidemiology
CHF caused by myocardial dysfunction is a serious malady that is a major cause of morbidity and mortality in children and adults. In these conditions, ventricular arrhythmias commonly occur and, in many instances, result in sudden death. These disorders are the most common diseases leading to cardiac transplantation, with an associated cost of billions of dollars annually in the United States. DCM is the most common cause of CHF, and, although the overall incidence varies, DCM is thought to occur in at least 40 of 100,000 population. The prevalence and incidence of DCM appear to be increasing. Depending on the diagnostic criteria used, the annual incidence varies between 5 and 8 cases per 100,000 population; the true incidence probably is underestimated by these figures because many asymptomatic cases go unrecognized. Nearly 5 million Americans have heart failure currently, with an increasing incidence with age. In individuals older than 65 years of age, the incidence approaches 10 per 1,000 population, and DCM accounts for more than 20% of all hospital admissions in this age group. In the pediatric population, newborns and infants have the highest rates of disease, with an annual incidence of 4.58 per 100,000 children (range 5.98 to 10.72 per 100,000). Symptomatic heart failure at all ages continues to confer a poor prognosis, with 1-year mortality rates still reported to be 45%.
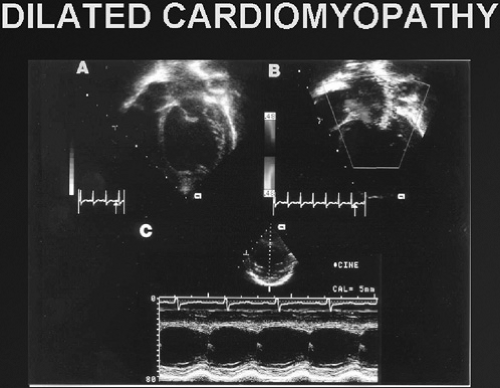 FIGURE 283.2. Echocardiography in dilated cardiomyopathy. A: Apical four-chamber view demonstrating a dilated left ventricle. B: Similar view showing mitral regurgitation. C: M-mode image showing poor systolic function. |
Clinical Manifestations
Idiopathic DCM is characterized by increased ventricular size (i.e., left ventricular or biventricular dilation) and reduced ventricular contractility (Fig. 283.2) in the absence of coronary artery disease, valvular abnormalities, or pericardial disease. Mitral regurgitation is a common finding, as are ventricular arrhythmias, particularly ventricular tachycardia (VT), torsade de pointes, and ventricular fibrillation. Clinical features include the signs and symptoms of CHF, which include breathlessness, fatigue, orthopnea, diaphoresis, chest pain, palpitations, exercise intolerance, and syncope. On physical examination, tachypnea, tachycardia, diaphoresis, an S3 or summation of S3 and S4 gallop, and hepatomagly are seen commonly with or without peripheral edema or ascites. Supportive data include radiographic evidence of cardiomegaly (with or without pulmonary edema) and reduced cardiac contractility with (or without) ventricular dilation on echocardiography. In adults, the condition is seen more frequently in men (2.5:1) and in African Americans (2.5:1), but the causes of these differences are not well understood. In children, it appears to be different, with the male-to-female ratio being 60% males, 40% females. The
clinical course of DCM, almost regardless of origin, may be progressive, with approximately 50% of individuals reported to die within 5 years of diagnosis without cardiac transplantation. The cause of death is divided evenly between sudden death and pump failure. Longer survival has been accomplished more recently with improved medical therapies (i.e., angiotensin-converting enzyme [ACE] inhibitors, beta blockers) and interventions (i.e., implantable defibrillators, ventricular assist devices).
clinical course of DCM, almost regardless of origin, may be progressive, with approximately 50% of individuals reported to die within 5 years of diagnosis without cardiac transplantation. The cause of death is divided evenly between sudden death and pump failure. Longer survival has been accomplished more recently with improved medical therapies (i.e., angiotensin-converting enzyme [ACE] inhibitors, beta blockers) and interventions (i.e., implantable defibrillators, ventricular assist devices).
Diagnostic Evaluation
The usual evaluation includes chest radiography, electrocardiography (ECG), echocardiography, and Holter monitoring. Exercise testing and cardiac catheterization, with or without endomyocardial biopsy (EMB), also may be performed. Blood and urine testing, particularly in young children, is mandatory to define an underlying cause.
Chest Radiography
Chest radiographs typically identify cardiomegaly and pulmonary edema with increased pulmonary vascular markings and Kerley B lines. In patients with a dilated left atrium, tracheal narrowing and atelectasis may occur. In some cases, pleural effusions are notable.
Electrocardiography
Sinus tachycardia occurs most commonly. In patients with a dilated left atrium, left atrial enlargement may occur. Ventricular arrhythmias and, on occasion, preexcitation may be identified, as may bundle branch block or atrioventricular block (AVB).
Echocardiography
Classic features include a dilated left ventricle with poor ventricular function. Doppler and color Doppler interrogation may demonstrate atrioventricular valve regurgitation (particularly mitral regurgitation). A pericardial effusion also may be seen, along with atrial dilation.
Holter Monitoring
The purpose of 24-hour ECG monitoring is to identify episodes of ventricular arrhythmias or bradycardia/AVB.
Cardiac Catheterization
Cardiac catheterization typically is not used as a diagnostic modality except to obtain EMB specimens to evaluate for possible myocarditis or, rarely, for mitochondrial abnormalities. Hemodynamic evaluation may identify elevated left ventricular end-diastolic pressure and pulmonary wedge pressures; in some cases, pulmonary vascular resistance may be elevated substantially. If endomyocardial biopsies are obtained, polymerase chain reaction (PCR) analysis for viral genome should be considered.
Metabolic Studies
In children, the underlying cause of DCM may include mitochondrial or metabolic derangement. For this reason, urine analysis for lactate, amino acids, and organic acid elevations may be useful and in some cases, such as Barth syndrome, may help to narrow the differential diagnosis. In Barth syndrome, for instance, elevated 3-methyl-glutaconic acid in a male patient with neutropenia would be diagnostic. Blood studies, including complete blood count, acylcarnitine profile (for fatty acid oxidation defects), pyruvate dehydrogenase complex, electrolyte profile, creatine kinase (with isoform analysis if elevated), plasma amino acids, and blood for genetic testing, all are reasonable. Elevation of creatine kinase muscle isoform (CK-MM) is useful to identify associated skeletal myopathy and may lead to identification of dystrophin (or other) mutations. Viral serologic studies may or may not be helpful. In some patients, particularly young children, skeletal muscle biopsy for microscopy, electron microscopy, and electron transport chain biochemistry can be useful and, if abnormal, should suggest the need for performing blood studies to evaluate for mitochondrial DNA mutations.
Arrhythmias and Conduction System Disease
The rate of sudden cardiac death in individuals with heart failure was reported by Stevenson and Stevenson to be six to nine times that seen in the general population. The basis of this sudden demise is thought to be the presence of ventricular tachyarrhythmias or severe bradyarrhythmias in children. The cause of these rhythm disturbances currently is unknown, but researchers have speculated that myocardial irritability caused by remodeling and fibrosis is a common inciting event. In addition, the myocardial conduction system is likely to be vulnerable to the same pathologic processes affecting the myocytes and interstitial elements, resulting in altered conduction. Furthermore, patients with DCM develop atrial stretching resulting from elevated end-diastolic ventricular pressure caused by poor systolic function and from mitral regurgitation, with electrical instability ensuing. In adults, this condition leads to atrial fibrillation, a decidedly uncommon event in children. However, abnormal myocardial conduction also may lead to ventricular conduction delays and bundle branch block, leading in some patients to worsening mechanical coupling, ventricular activation and contraction, ventricular dyssynchony, abnormal diastolic function, and significant hemodynamic consequences that could trigger ventricular arrhythmias. These arrhythmias dramatically increase the risk of sudden death and currently are treated with internal defibrillators and/or antiarrhythmic drugs. In adults, left bundle branch block is a significant predictor of sudden death and a common finding in adults with heart failure; however, this does not appear to be the case in children.
Heart Failure with Preserved Systolic Function
The usual teaching regarding heart failure specifies the need for either systolic dysfunction (pump failure) or volume overload. However, another form of heart failure has become increasingly common in adults (20% to 50% of patients with heart failure) and is beginning to crop up in a small number of children with heart failure. In this patient cohort, the usual systolic function indices (shortening fraction, ejection fraction) are preserved. Despite normal contractility, diastole (relaxation) is impaired, and cardiac output is therefore limited by the abnormal filling characteristics of the ventricles, particularly during exercise. For a given ventricular volume, ventricular pressures are elevated, leading to pulmonary congestion, dyspnea, and edema. These symptoms are identical to those of patients with pump failure. This disorder is an unusual occurrence in children and is seen most commonly in elderly, usually female, patients with obesity, hypertension, and diabetes. Mortality is similar to that of typical CHF.
Syndrome of Heart Failure
Traditionally, the syndrome of heart failure has been viewed as a constellation of clinical findings resulting from inadequate systolic function (“pump function”). However, since the early 1990s, this view has been altered by a variety of clinical and basic data, including the continued poor outcomes of patients despite treatment with therapies designed to improve systolic function. In addition, information regarding inflammatory mediators, apoptosis, structure-function studies, and genetics has resulted in a concept that the syndrome of heart failure occurs as a result of a complex interaction of structural, functional, and biologic disturbances of the heart.
The current concepts regarding the syndrome of heart failure integrate a series of models of heart failure, including the hemodynamic model, neurohormonal model, structural model, and autocrine-paracrine model of heart failure. In the hemodynamic model, alterations in load on the failing ventricle are central to the hypothesis, which relies heavily on pump dysfunction
as the key contributor. Based on this model, therapies were developed that focused on inotropic agents and vasodilators, with a goal of improving contractility. In distinction to this model, the neurohormonal model focused on the importance of the renin-angiotensin-aldosterone system and the sympathetic nervous system as central to the development of heart failure and resulted in attempts to pharmacologically antagonize the effect of circulating norepinephrine and angiotensin II. The autocrine-paracrine model was developed because of the findings that norepinephrine, angiotensin II, and other vasoactive substances such as brain natriuretic peptide (BNP) also are synthesized within the myocardium, thereby having actions in an autocrine and paracrine manner in addition to their actions in the circulation. Finally, the structural model focuses on the necessity of proper interactions between the sarcomere and sarcolemma via cytoskeletal and other proteins. These interactions, as well as models relying on inflammatory mediators and apoptosis, all have been used to explain the features of heart failure; likely each plays a role in the clinical disorder.
as the key contributor. Based on this model, therapies were developed that focused on inotropic agents and vasodilators, with a goal of improving contractility. In distinction to this model, the neurohormonal model focused on the importance of the renin-angiotensin-aldosterone system and the sympathetic nervous system as central to the development of heart failure and resulted in attempts to pharmacologically antagonize the effect of circulating norepinephrine and angiotensin II. The autocrine-paracrine model was developed because of the findings that norepinephrine, angiotensin II, and other vasoactive substances such as brain natriuretic peptide (BNP) also are synthesized within the myocardium, thereby having actions in an autocrine and paracrine manner in addition to their actions in the circulation. Finally, the structural model focuses on the necessity of proper interactions between the sarcomere and sarcolemma via cytoskeletal and other proteins. These interactions, as well as models relying on inflammatory mediators and apoptosis, all have been used to explain the features of heart failure; likely each plays a role in the clinical disorder.
Despite the probable complexity of interactions resulting in heart failure, a major abnormality at the center of the disorder and its reversal is the process of remodeling. Improved outcomes appear to be linked to the ability to reverse this process (“reverse-remodeling”). Remodeling of the left ventricle is a process in which ventricular size, shape, and function are altered because of mechanical, genetic, and neurohormonal factors that lead to hypertrophy, myocyte loss, and interstitial fibrosis. In DCM, the process of progressive ventricular dilation and morphologic ventricular change to a more spheric shape, associated with changes in ventricular function and/or hypertrophy, occurs without known initiating disturbance except in patients with myocardial infarction. Because of this remodeling event, mitral regurgitation may develop as the geometric relations among the mitral valve apparatus, mitral ring, and papillary muscles are altered. The presence of mitral regurgitation results in increasing volume overload on an already compromised ventricle, further contributing to remodeling, symptoms, progression of disease, and left atrial dilation.
Clinical Genetics
DCM initially was thought to be inherited in a small percentage of cases until Michels and associates showed that approximately 20% of probands had family members with echocardiographic evidence of DCM when family screening was performed. More recently, inherited, familial DCM (FDCM) has been shown to occur in more than 30% of cases. In the remaining cases, many are acquired, with viral myocarditis playing a significant role.
As noted, more than 30% of patients with DCM have a familial form of disease. Autosomal dominant inheritance is the predominant pattern of transmission, with X-linked, autosomal recessive, and mitochondrial inheritance being less common. Mitochondrial inheritance is seen most commonly in childhood forms of FDCM, whereas X-linked and autosomal recessive forms probably are evenly mixed between childhood and adult forms of disease. During the past decade, progress has been made in the understanding of the genetic origin of FDCM. Initial progress was made studying families with X-linked forms of DCM, with the autosomal dominant forms of DCM beginning to unravel during the past few years. Box 283.2 gives additional information concerning the genetics of DCM.
Muscle Is Muscle: Cardiomyopathy and Skeletal Myopathy Genes Overlap
Nearly all the genes identified for inherited DCM also are known to cause skeletal myopathy in humans and/or mouse models. In the case of dystrophin, mutations cause Duchenne muscular dystrophy and Becker muscular dystrophy, whereas delta-sarcoglycan mutations cause LGMD2F. Lamin A/C has been shown to cause autosomal dominant Emery-Dreifuss muscular dystrophy and LGMD1B, whereas actin mutations are associated with nemaline myopathy. Desmin, G4.5, alpha-dystrobrevin, Cypher/ZASP, MLP, alpha-actinin-2, titin, and beta-sarcoglycan mutations also have associated skeletal myopathy, a finding suggesting that cardiac and skeletal muscle function is interrelated and that possibly the skeletal muscle fatigue seen in patients with DCM with and without CHF may result from primary skeletal muscle disease and not solely cardiac dysfunction. It also suggests that the function of these muscles has a “final common pathway,” and cardiologists and neurologists should consider evaluation of both sets of muscles.
Further support for this concept comes from studies of animal models. Mutations in delta-sarcoglycan in hamsters result in cardiomyopathy, and mutations in all sarcoglycan subcomplex genes in mice cause skeletal and cardiac muscle disease. Mutations in other DAPC genes as well as dystrophin in murine models also consistently demonstrate abnormalities of skeletal and cardiac muscle function. Arber and associates also produced a mouse deficient in muscle MLP, a structural protein that links the actin cytoskeleton to the contractile apparatus. The resultant mice develop severe DCM, CHF, and disruption of cardiac myocyte cytoskeletal architecture. Murine mutations in titin, cypher, alpha-dystrobrevin, desmin, and other all demonstrate cardiac and skeletal muscle disease. Finally, Badorff and colleagues showed that DCM that develops after viral myocarditis has a mechanism similar to that of the inherited forms. Using coxsackievirus B3 infection of mice, the authors showed that the coxsackievirus B3 genome encodes for a protease (enteroviral protease 2A) that cleaves dystrophin at the third hinge region of dystrophin, resulting in force transmission abnormalities and DCM. In addition, Xiong and associates showed that abnormal dystrophin increases susceptibility to viral infection and resultant myocarditis. As reported previously by our laboratory, a similar dystrophin mutation affects the first hinge region of dystrophin in patients with X-linked cardiomyopathy, demonstrating a consistent mechanism of DCM development, abnormalities of the cytoskeleton/ sarcolemma, and sarcomere. In addition, we have shown that N-terminal dystrophin is reduced or absent in hearts of patients with all forms of DCM (ischemic, acquired, genetic, idiopathic) and that reduction of mechanical stress by use of left ventricular assist devices (LVADs) results in reverse remodeling of dystrophin and of the heart itself.
Treatment
The therapy for DCM has changed since the early 1990s from focusing on improving systolic function to improving cardiac efficiency. In the past, inotropic therapy was a mainstay of therapy, whereas today it is less popular. We tend to avoid using inotropic medications as much as possible and instead use combinations of medication aimed at avoiding arrhythmias. Low-dose (renal dose) dopamine, in addition to milrinone and diuretics, has been useful. More recently, neseritide, a BNP synthetic, has been useful in improving urine output and cardiac function. In children requiring intubation and mechanical ventilation, calcium infusion with or without vasopression has worked well in selected patients. In some centers, dobutamine and epinephrine remain mainstays of treatment.
In patients in whom these therapies are ineffective, mechanical assist device therapies using an LVAD device or extracorporeal membrane oxygenator (ECMO) has been lifesaving. We have hypothesized that LVAD treatment reduces mechanical stress on the myocardium, thus enabling reverse remodeling via dystrophin to occur. We previously showed that the
N-terminus of dystrophin is lost in DCM, and, after weeks of LVAD treatment, reversal occurs, resulting in relinkage of the sarcolemma and sarcomere at the actin-binding domain of dystrophin. More recently, we demonstrated similar findings in the right ventricle as well and showed biventricular reversal using either pulsatile or continuous-flow LVADs. In patients well enough to be treated as outpatients, ACE inhibitors and beta blockers have been shown to be efficacious. The use of diuretics also may be useful. The certainty of efficacy of digoxin has come into question, and controversy also exists regarding the use of “vitamins” such as coenzyme Q10, carnitine, thiamin, riboflavin, and others. In the latter case, these medications usually are reserved for patients with mitochondrial or metabolic-based disease.
N-terminus of dystrophin is lost in DCM, and, after weeks of LVAD treatment, reversal occurs, resulting in relinkage of the sarcolemma and sarcomere at the actin-binding domain of dystrophin. More recently, we demonstrated similar findings in the right ventricle as well and showed biventricular reversal using either pulsatile or continuous-flow LVADs. In patients well enough to be treated as outpatients, ACE inhibitors and beta blockers have been shown to be efficacious. The use of diuretics also may be useful. The certainty of efficacy of digoxin has come into question, and controversy also exists regarding the use of “vitamins” such as coenzyme Q10, carnitine, thiamin, riboflavin, and others. In the latter case, these medications usually are reserved for patients with mitochondrial or metabolic-based disease.
BOX 283.2. Genetics of Dilated Cardiomyopathy
X-Linked Cardiomyopathies
X-Linked Dilated Cardiomyopathy
X-linked dilated cardiomyopathy (XLCM) was described first in 1987 by Berko and Swift as DCM occurring in male patients in the teen years and early 20s with rapid progression from congestive heart failure (CHF) to death resulting from ventricular tachycardia/ventricular fibrillation (VT/VF) or cardiac transplantation. Patients with this condition are distinguished by elevated serum CK-MM. Female carriers tend to develop mild to moderate DCM in the fifth decade of life, and the disease is slowly progressive. In 1993, Towbin and colleagues were the first to identify the disease-causing gene and to characterize the functional defect. In this report, the dystrophin gene was shown to be responsible for the clinical abnormalities, and protein analysis by immunoblotting demonstrated severe reduction or absence of dystrophin protein in the heart of these patients. These findings were confirmed later by Muntoni and associates when a mutation in the muscle promoter and exon 1 of dystrophin was identified in another family with XLCM. Subsequently, multiple mutations have been identified in dystrophin in patients with XLCM.
Dystrophin is a cytoskeletal protein that provides structural support to the myocyte by creating a lattice-like network to the sarcolemma. In addition, dystrophin plays a major role in linking the sarcomeric contractile apparatus to the sarcolemma and extracellar matrix. Furthermore, dystrophin is involved in cell signaling, particularly through its interactions with nitric oxide synthase. When mutated, the dystrophin gene also is responsible for Duchenne and Becker muscular dystrophy (DMD/BMD). These skeletal myopathies present early in life (DMD is diagnosed in patients younger than age 12 years, whereas BMD is seen in male teenagers older than 16 years of age) and most patients develop DCM before reaching their twenty-fifth birthday. In most patients, CK-MM is elevated similar to that seen in XLCM; in addition, manifesting female carriers develop disease late in life, similar to those with XLCM. Furthermore, immunohistochemical analysis demonstrates reduced levels (or an absence) of dystrophin, similar to that seen in the hearts of patients with XLCM.
Murine models of dystrophin deficiency demonstrate abnormalities of muscle physiology based on membrane structural support abnormalities. In addition to the dysfunction of dystrophin, mutations in dystrophin secondarily affect proteins that interact with dystrophin. At the amino-terminus (N-terminus), dystrophin binds to the sarcomeric protein actin, a member of the thin filament of the contractile apparatus. At the carboxy-terminus (C-terminus), dystrophin interacts with alpha-dystroglycan, a dystrophin-associated membrane-bound protein that is involved in the function of the dystrophin-associated protein complex (DAPC), which includes beta-dystroglycan, the sarcoglycan subcomplex (alpha, beta, gamma, delta, and ε sarcoglycan), syntrophins, and dystrobrevins (see Fig. 283.1). In turn, this complex interacts with alpha-2-laminin and the extracellular matrix. As with dystrophin, mutations in these genes lead to muscular dystrophies with or without cardiomyopathy, a finding supporting the contention that these proteins are important to the normal function of the myocytes of the heart and skeletal muscles. In both cases, mechanical stress appears to play a significant role in the age-onset dependent dysfunction of these muscles. The information gained from the studies on XLCM, DMD, and BMD led us to hypothesize that DCM is a disease of the cytoskeleton/sarcolemma that affects the sarcomere. We also have suggested that dystrophin mutations play a role in idiopathic DCM in male patients. Recently, we showed that 3 of 22 boys with DCM studied for dystrophin mutations using a rapid DNA mutation screening method had mutations and all had elevated CK-MM as well. In addition, eight families with DCM and possible X-linked inheritance also were screened; in three of eight families, dystrophin mutations were noted. Again, CK-MM was elevated in all subjects carrying mutations.
Barth Syndrome
Initially described as X-linked cardioskeletal myopathy with abnormal mitochondria and neutropenia by Neustein and Barth and their colleagues, this disorder typically presents in male infants as CHF associated with neutropenia (cyclic) and 3-methylglutaconic aciduria. Mitochondrial dysfunction is noted on electron microscope and electron transport chain biochemical analysis. Recently, abnormalities in cardiolipin have been noted. Echocardiographically, these infants typically have left ventricular dysfunction with left ventricular dilation, endocardial fibroelastosis, or a dilated hypertrophic left ventricle. In some cases, these infants die of CHF/sudden death VT/VF, or sepsis caused by leukocyte dysfunction. Most of these children survive infancy and do well clinically, although DCM usually persists. In some cases, cardiac transplantation has been performed. Histopathologic evaluation typically demonstrates the features of DCM, although endocardial fibroelastosis may be prominent, and the mitochondria are abnormal in shape and abundance.
The genetic basis of Barth syndrome was described first by Bione and colleagues, who cloned the disease-causing gene, G4.5. This gene encodes a novel protein called tafazzin, whose function is not currently known. Researchers have speculated, however, that the gene product is an acyltransferase based on the cardiolipin abnormalities. Mutations in G4.5 result in a wide clinical spectrum, which includes apparent classic DCM, hypertrophic DCM, endocardial fibroelastosis, or left ventricular noncompaction (LVNC). In the latter case, the LVNC is characterized by deep trabeculations giving the appearance of a “spongiform” myocardium (see Fig. 283.11). The mechanisms responsible for this clinical heterogeneity are not known. More detail is provided regarding LVNC in the section on this disorder.
Autosomal Dominant Dilated Cardiomyopathy
The most common form of inherited DCM is the autosomal dominant form of disease. These patients present as classic “pure” DCM or DCM associated with conduction system disease (CDDC). In the latter case, patients usually present in their 20s with mild CDDC that can progress over the course of decades to complete heart block. DCM usually presents late in the course but is out of proportion to the degree of CDDC. The echocardiographic and histologic findings in both subgroups are classic for DCM, although the conduction system may be fibrotic in patients with CDDC. In both groups of patients with DCM, VT, VF, and torsade de pointes occur and may result in sudden death.
Genetic heterogeneity exists for autosomal dominant DCM, with 15 loci mapped for pure DCM and 5 loci for CDDC. In the case of pure DCM, 10 genes have been identified to date and include 3 by our group (delta-sarcoglycan, alpha-actinin-2, ZASP), as well as actin, desmin, troponin T, beta-myosin heavy chain, titin, metavinculin, myosin binding protein C, alpha-tropomyosin, MLP, and phospholamban (see Table 283.1).
Most of the genes identified to date encode either cytoskeletal or sarcomeric proteins. In the case of cytoskeletal proteins (desmin, delta-sarcoglycan, metavinculin, MLP), defects of force transmission are considered to result in the DCM phenotype, whereas defects of force generation have been speculated to cause sarcomeric protein-induced DCM.
Cardiac actin is a sarcomeric protein that is part of the sarcomeric thin filament interacting with tropomyosin and the troponin complex. As previously noted, actin plays a significant role in linking the sarcomere to the sarcolemma via its binding to the N-terminus of dystrophin. Interestingly, the mutations in actin that resulted in DCM as described by Olson and associates appear to be involved directly in the binding of dystrophin, whereas mutations in the sarcomeric end of actin result in hypertrophic cardiomyopathy (HCM). The DCM-causing mutations are thought to result in DCM by causing force transmission abnormalities. Further, actin interacts in the sarcomere with TnT and beta-MHc, two other genes resulting in either DCM or HCM depending on the position of the mutation. In the case of TnT and beta-MHC, force generation abnormalities have been speculated as the responsible mechanism.
Desmin is a cytoskeletal protein that forms intermediate filaments specific for muscle. This muscle-specific 53-kDa subunit of class III intermediate filaments forms connections between the nuclear and plasma membranes of cardiac, skeletal, and smooth muscle. Desmin is found at the Z-lines and intercalated disc of muscle; its role in muscle function appears to involve attachment or stabilization of the sarcomere. Mutations in this gene appear to cause abnormalities of force and signal transmission similar to those thought to occur with actin mutations.
Another DCM-causing gene, delta-sarcoglycan, encodes a member of the sarcoglycan subcomplex of the DAPC. This gene encodes for a protein involved in stabilization of the myocyte sarcolemma, as well as signal transduction. Mutations identified in familial and sporadic cases resulted in reduction of the protein within the myocardium. In the absence of delta-sarcoglycan, the remaining sarcoglycans (delta, beta, gamma, epsilon) cannot assemble properly in the endoplasmic reticulum. Mouse models of delta-sarcoglycan deficiency demonstrate dilated, HCM, sarcolemmal fragility, and disrupted vasculin smooth muscle that leads to vascular spasm, including coronary spasm. In addition, mutations in this gene lead to the phenotype of the cardiomyopathic Syrian hamster. Other human mutations in delta-sarcoglycan cause a form of autosomal recessive limb girdle muscular dystrophy (LGMD2F) that rarely is associated with heart disease.
The final cytoskeletal protein-encoding gene, metavinculin, encodes vinculin and its splice variant metavinculin. Vinculin is expressed ubiquitously, and metavinculin is coexpressed with vinculin in heart, skeletal, and smooth muscle, with this protein complex localized to subsarcolemmal costameres in the heart, where they interact with alpha-actinin, talin, and gamma-actin to form a microfilamentous network linking cytoskeleton and sarcolemma. In addition, these proteins are present in adherens junctions in intercalated discs and participate in cell-cell adhesion. Mutations in metavinculin have been shown to disrupt the intercalated discs and alter actin filament cross-linking.
As previously noted, mutations in the sarcomere may produce HCM or DCM. In the latter case, abnormalities in force generation or transmission are thought to contribute to the development of this phenotype. In addition to mutations in the thin filament protein actin, mutations in the thick filament protein-encoding gene beta-myosin heavy chain have been shown to cause DCM with associated sudden death in at least one infant, as well as DCM in older children and adults. Mutations in this gene are thought to perturb the actin-myosin interaction and force generation or alter cross-bridge movement during contraction. Mutations in cardiac troponin T, a thin filament protein, have been speculated to disrupt calcium-sensitive troponin C binding. Mutations in phospholamban also have been identified that further support calcium handling as a potentially important mechanism in the development of DCM. Interestingly, Haghighi and associates identified homozygous mutations causing DCM and heart failure, whereas heterozygoes had cardiac hypertrophy. Recessive mutation in troponin I is thought to impair the interaction with troponin T. alpha-Tropomyosin mutations also have been identified and were predicted to alter the surface charge of the protein leading to impaired interaction with actin.
A recent area of interest for evaluation at the molecular level is the Z-disc. Knoll and colleagues identified mutations in muscle MLP and demonstrated that these mutations result in defects in the interaction with telethonin. Using mouse models, these investigators also demonstrated that MLP acts as a stretch sensor and that mutant MLP causes defects in this activity. More recently, Mohapatra and associates demonstrated mutations in MLP in families and sporadic cases and identified abnormalities in the T-tubule system and Z-disc architecture by electron microscopy, which correlates with the histopathology seen in MLP-knockout mice. This finding was supported further by the finding of reduced expression of MLP in chronic human heart failure. In addition, mutations in alpha-actinin-2, which is involved in crosslinking actin filaments and shares a common actin binding domain with dystrophin, also were identified in FDCM, which disrupts its binding to MLP. Finally, Vatta and colleagues identified mutations in the Z-band alternatively spliced PDZ-motif protein ZASP, the human homolog of the mouse cypher gene, which when disrupted leads to DCM. Multiple mutations in this gene were identified in families and sporadic cases of DCM and with LVNC. This protein, which interacts with alpha-actinin-2, disrupts the actin cytoskeleton when mutated. Another gene, titin, which encodes the giant sarcomeric cytoskeletal protein titin that contributes to the maintenance of the sarcomere organization and myofibrillar elasticity, interacts with these proteins at the Z-disc/I-band transition zone. Mutations have been identified in FDCM as well.
As seen in pure autosomal dominant DCM, genetic heterogeneity also exists for CDDC. To date, CDDC genes have been mapped to chromosomes 1p1-1q1, 2q14-21, 3p25-22, and 6q23. The only gene thus far identified was reported initially by Fatkin and colleagues and Brodsky and associates to be lamin A/C on chromosome 1q21, which encodes a nuclear envelope intermediate filament protein.
The lamins are located in the nuclear lamina at the nucleoplasmic side of the inner nuclear membrane, and lamin A and C are expressed in heart and skeletal muscle. Mutations in this gene initially were reported to cause the autosomal dominant form of Emery-Dreifuss muscular dystrophy, which has skeletal myopathy associated with DCM and conduction system disease. It also has been found to cause a form of autosomal dominant limb girdle muscular dystrophy (LGMD1B), which also is associated with conduction system disease. Multiple mutations have been identified in patients with DCM and conduction system disease, which, in some patients, had mildly elevated CK. This gene defect appears to be a relatively common occurrence in patients with CDDC. The mechanisms responsible for the development of DCM and conduction system abnormalities are unknown.
Other novel approaches currently undergoing investigation include biventricular pacing (also known as resyndronization therapy). In those patients with AVB, a pacemaker also is appropriate, whereas patients with VT currently are considered for use of internal defibrillators.
Myocarditis
Myocarditis is a process characterized by inflammatory infiltrate of the myocardium with necrosis and/or degeneration of adjacent myocytes not typical of the ischemic damage associated with coronary artery disease. This definition does not account for the underlying cause.
Etiology
Most cases of myocarditis in the United States and Western Europe result from viral infections. The most common viral causes include adenovirus (particularly serotypes 2 and 5) and enterovirus (coxsackieviruses A and B, echovirus, poliovirus), particularly coxsackievirus B (Table 283.2). However, many other viral causes of myocarditis in children and adults, including influenza, cytomegalovirus, herpes simplex virus, parvovirus, hepatitis C, rubella, varicella, mumps, EBV, HIV, and respiratory syncytial virus, among others, have been described. Other nonviral causes include the following: other infectious agents such as rickettsiae, bacteria, protozoa and other parasites, fungi, and yeasts (Table 283.3); various drugs including antimicrobial medications; hypersensitivity, autoimmune, or collagen-vascular diseases such as systemic lupus erythematosus, mixed connective tissue disease, rheumatic fever, rheumatoid arthritis, and scleroderma; toxic reactions to infectious agents (e.g., diphtheria); or other disorders such as Kawasaki disease and sarcoidosis (see Table 283.3). In most cases, however, “idiopathic” myocarditis is encountered.
Epidemiology
Myocarditis is a disorder that is underdiagnosed, but the incidence of the usual lymphocytic form of myocarditis has been reported to be from 4% to 5% (as obtained from reports of young men dying of trauma) to as high as 16% to 21% (as found
in autopsy series of children dying suddenly). In adults with unexplained DCM, the reported incidence varies between 3% and 63%, although the large multicenter Myocarditis Treatment Trial, which was strictly based on the “Dallas criteria,” reported a 9% incidence.
in autopsy series of children dying suddenly). In adults with unexplained DCM, the reported incidence varies between 3% and 63%, although the large multicenter Myocarditis Treatment Trial, which was strictly based on the “Dallas criteria,” reported a 9% incidence.
TABLE 283.2. VIRAL POLYMERASE CHAIN REACTION (PCR) ANALYSIS OF MYOCARDITIS AND DILATED CARDIOMYOPATHY (DCM): THE DETECTION OF VIRUSES BY PCR IN MYOCARDIAL SAMPLES | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 283.3. CAUSES OF MYOCARDITIS | |
|---|---|
|
Usually sporadic, viral myocarditis also can occur as an epidemic. Epidemics usually are seen in newborns, most commonly in association with coxsackievirus B. Intrauterine myocarditis also has been seen during epidemics, as well as sporadically. Postnatal spread of coxsackievirus is via the fecal-oral or airborne route. The WHO reports that this ubiquitous family of viruses results in cardiovascular sequelae in less than 1% of infections, although this incidence increases to 4% when coxsackievirus B3 is considered. Other important viral causes, such as adenovirus and influenza A, are transmitted through the air. Although the disease can occur equally throughout the year, the exact cause probably is season-dependent [in other words, certain viral causes are seasonal (e.g., coxsackievirus) whereas others are year-round conditions (e.g., adenovirus)].
Clinical Manifestations
Differences in presentation are seen depending on the age of the child (i.e., newborn or infant versus child or adolescent), thus rendering establishing the diagnosis challenging. Adults present with findings similar to those of adolescents. In general, myocarditis should be considered in all children and adults with new-onset CHF in whom no other cause is found. In many cases, an antecedent, nonspecific flu-like illness or episode of gastroenteritis may precede the manifestation of symptoms of CHF. The younger the child, the poorer the prognosis, with death reported in more than 50% of neonates and infants (18 months or younger). In a study by Bowles and colleagues, 81% of newborns (78 of 93) with symptomatic myocarditis referred for molecular diagnosis died, with death occurring in 71% (108 of 152) of infants between 1 month and 12 months of age and in 58% (11 of 18) of those 12 to 18 months of age.
Newborns and Infants
Newborns or infants typically present with fever, irritability or listlessness, periodic episodes of pallor (which may precede the sudden onset of cardiorespiratory symptoms including tachypnea or respiratory distress), and diaphoresis. Poor appetite or vomiting also can be seen frequently. Sudden death may occur in this subgroup of children. On physical examination, pallor and mild cyanosis commonly are noted. The skin usually is cool and mottled (and sometimes clammy), consistent with poor perfusion resulting from decreased cardiac output. Respirations usually are rapid and labored; grunting may be prominent, but rales are uncommon findings (in fact, if rales are auscultated, one should strongly consider pneumonia with or without sepsis as the diagnosis). The cardiovascular examination is consistent with CHF and includes resting tachycardia and a gallop rhythm, muffled heart sounds, and frequently an apical systolic murmur caused by mitral regurgitation. In some of these young children, particularly newborns, a tricuspid regurgitation murmur may also be identified. The pulses are usually thready, and hepatomegaly is usually obvious. Depending on the underlying origin, splenomegaly also may be prominent, but it is very uncommon when myocarditis is the cause of heart failure. Arrhythmias (supraventricular tachycardia or VT) or AVB also may occur.
An important point is that the younger the child, the more likely that intrauterine myocarditis occurred and that the findings may be associated more with chronic disease than otherwise expected in acute disease. In addition, as noted, this patient group has the worst prognosis.
Children, Adolescents, and Adults
Older children, adolescents, and adults commonly report a recent history of viral disease, generally 10 to 14 days before presentation. Initial symptoms include lethargy, low-grade fever, and pallor; the child usually has decreased appetite and may complain of abdominal pain. Diaphoresis, palpitations, rashes, intolerance for exercise, and general malaise are common signs and symptoms. Later in the course of illness, respiratory symptoms become predominant; syncope or sudden death may occur as a result of cardiac collapse. Findings on physical examination are consistent with CHF. Unlike in newborns, jugular venous distention and pulmonary rales may be found, and resting tachycardia may be prominent. Arrhythmias, including atrial fibrillation, supraventricular tachycardia, or VT, as well as AVB, may occur.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


