Key Points
- •
Each crystal type associated with arthritis forms in specific joint tissues dependent on the distinct metabolic environment.
- •
Crystals require additional factors beyond tissue deposition to incite inflammation.
- •
Crystals within cartilage can lead to degenerative changes resulting from both metabolic and physical effects.
- •
Monosodium urate monohydrate (MSU) crystals have the potential to precipitate in loose connective tissues of joints and bursae.
- •
Calcium pyrophosphate dihydrate crystal deposition is restricted to hyaline cartilage, fibrocartilage, and fibrous tissue environments. Acute arthritis is believed to be triggered by crystal shedding from cartilage into the joint space.
- •
Basic calcium phosphate crystals within synovial fluid may be derived from bone and calcified cartilage fragments exposed to the articular surface, or from periarticular or bursal basic calcium phosphate deposition.
General Pathology of Crystal-Associated Joint Disease
It is fascinating to consider that almost all the advances recorded in this chapter stem from the pioneering studies half a century ago of Zitnan and colleagues on chondrocalcinosis and of McCarty and colleagues on the detection and separation of calcium pyrophosphate dihydrate (CPPD) crystals from monosodium urate (MSU) crystals in synovial fluid (SF).
Common to the pathogenesis of crystal-associated arthropathy is a local tissue metabolic environment that is supersaturated with ions of at least one of the crystal components and an event or series of events that facilitates crystal precipitation from a supersaturated state ( Fig. 1-1 ). Regardless of systemic metabolic abnormalities or systemic ion saturation, the supersaturated metabolic environment must be local to the tissues in which the crystals deposit: elevated tissue urate concentration (urate) in gout, elevated tissue inorganic pyrophosphate ion concentration (PP i ) associated with CPPD crystal deposition, and increased inorganic phosphate ion concentration (P i ) associated with basic calcium phosphate (BCP) crystal formation.
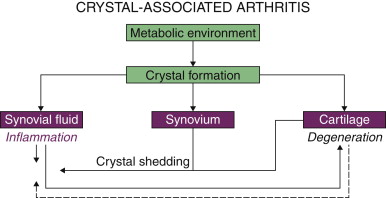
Under normal conditions, ions can remain in a supersaturated state until an environmental event occurs that renders the ions less soluble and promotes crystal nucleation. This event may be the increased kinetic energy of sudden mechanical force or increased free ion concentration associated with tissue dehydration, pH change, decreased temperature, enzymatic activity, concentration change in secondary ions, or absorption of ions on solid particles. This results in initial formation of molecular clusters (nuclei). From molecular clusters, depending on relative molecular solubilities between larger particles and smaller ones, as well as molecular diffusion rate, crystals rapidly grow to sizes specific to each crystal phase (Ostwald ripening factor). Under some conditions, new crystals grow on preexisting crystals (epitaxial growth). Crystals elongate on the face with the highest energy. Adhesion of proteins and other substances has crystal face specificity that can be disrupted by local ionic conditions. With crystal formation, there is local tissue depletion of the contributing ions as they are incorporated into the crystals. In gout, this shift in equilibrium brings in more urate from systemic interstitial fluid, stabilizing and amplifying the crystal deposition; in CPPD deposition disease, the local cartilage or tendon tissue matrix may remain depleted of PP i .
The biologic reactions that follow depend not only on crystal size and charge but also on the tissue environment in which crystals are deposited. In synovial or bursal spaces including the synovial spaces associated with tendons, acute inflammation can occur. In cartilage or fibrous tissue environments such as tendon insertions or intervertebral disc, slower, more chronic reactions to the crystals and/or the crystal-associated inflammation lead to impaired functionality of the tissue (degeneration) (see Fig. 1-1 ).
Crystal formation does not necessarily lead to immediate tissue reaction. When crystals form in connective tissues, the crystal surfaces rapidly become covered with matrix material, usually hyaluronate proteoglycan, present in the tissue space. Proteoglycan has at least two functions related to crystal reactions. First, intact proteoglycan increases hydration in the local environment via coordination of water with its negative charge groups; second, proteoglycan provides a barrier to cells by physical gelation. To incite inflammation in synovial spaces, it has been suggested that crystals must first become coated (opsonized) with proteins that promote cell adhesion or inflammatory response, such as IgG, or that urate crystals must shed surface proteins that inhibit cell interaction such as low-density lipoprotein apolipoprotein B (see Chapter 5 ). This occurs when there is increased vascular permeability to proteins adjacent to the space where crystals precipitate. This may be related to low-grade inflammation from other causes or from rapid crystal precipitation, which by itself may induce increased vascular permeability, perhaps via pH change. However, if crystals form slowly or if the SF is particularly viscous, the crystals could become coated with sufficient proteoglycan to act as a barrier to both acute reactant proteins and inflammatory cells.
Therefore, without inducing acute inflammation, crystals can be present in the synovial space, in gout, even producing a thick layer on the cartilage surface ( Fig. 1-2 ), which is also detectable by high-resolution ultrasound in gout. Similarly, quiescence of crystal-induced inflammation (see Chapter 5 ) may well involve large proteoglycans that separate crystals from cells.

Although the amount of inflammation varies with crystal type, crystal aggregation, the volume and rapidity of crystal precipitation or shedding, and innate immune inflammatory mechanisms are common to both urate and CPPD crystals, with BCP crystals having multiple distinctions, resulting in a less inflammatory picture in the joint space (see Chapter 6 , Chapter 21 , Chapter 23 ). The acute inflammatory mechanisms in crystal-associated arthritis have been documented extensively using morphologic, biochemical, cell biology, and molecular biologic techniques. This is of great interest for understanding inflammation biology and its control by drugs. In fact, as pathologic agents to study inflammation mechanisms, crystals are ideal because their composition and surfaces are known in precise atomic detail. Further, for CPPD in particular but also for other pathologic crystals, the crystal size, shape, and surface can be modified experimentally to help understand the effect of each parameter of the inflammatory stimulus.
Briefly, under the appropriate conditions described earlier, crystals precipitate or, in the crystal “autoinjection” model, free non-aggregated crystals are shed into loci in the joint where synovial lining cells and resident phagocytes are able to interact with the crystals, With changes in vascular permeability, the crystal surfaces become coated with blood-derived proteins. As reviewed in Chapter 5 , for gout, products of resident cells, such as mast cells, synovial lining cells, macrophages, and complement activation are major factors in attracting blood monocytes and neutrophils with phagocytosis capability to the joint. The phagocytes bind to the crystal surface and, recognizing the crystal as a foreign substance, phagocytose or, with larger crystals such as urate or CPPD, attempt to phagocytose the crystals. Amplification of inflammation ensues, with monocyte and neutrophil recruitment, phagocyte activation, with elaboration of increased lysosomes within the cell, focal cell membranolysis facilitated by crystal physical properties, incorporation of the crystal within the phagosome, merging of the lysososome with the phagosome, as well as, in macrophage lineage cells, activation of the NLRP3 inflammasome.
From a pathology perspective, because the crystals are relatively insoluble, this process is often not successful, resulting in increased cell membrane permeability, with consequent cell death and fragmentation. The released lysosomal enzymes bind to extracellular crystals and, along with hydrogen peroxide, facilitate crystal dissolution. The cell debris and released intracellular enzymes act to attract more phagocytes and increase vascular congestion and permeability, further resulting in acute inflammation characterized clinically by the familiar characteristics of joint pain, swelling, redness, warmth, and loss of function. After the neutrophils and monocytes are recruited into the space containing the crystals and monocytes differentiate to mature macrophages, over time, usually a few days, the extracellular enzymes and reactive oxygen species break down the inflammatory proteins ; the crystals are cleared by phagocytosis followed by crystal dissolution within the cell phagolysosomes, by crystal dissolution in the extracellullar space facilitated by increased solute (synovial effusion), pH change, and, for CPPD at least, extracellular enzymes. Under appropriate conditions, crystals become sequestered from the inflammatory cells by hyaluronate and proteoglycans, newly secreted from synovial cells.
In the usual circumstance, crystal-associated inflammation resolves completely with substantial clearance of crystals from the joint space (although not typically complete in patients with gout as a prime example), and without residual chronic, clinical inflammation or synovial effusion. Eliseo Pascual and colleagues have observed that SF leukocyte counts can remain mildly elevated relative to normal joints in patients with gout. 15 Nevertheless, the synovial tissue structure is usually restored to its state before the acute inflammation. This means that the synovial cell proliferation and fibrosis in crystal arthropathy are associated with persistence of crystals or have been caused by other, indirectly crystal-related pathology.
When crystals form in dense connective tissue rich in proteoglycans and collagen, it has been thought that the crystals are inaccessible to proinflammatory proteins, neutrophils, and macrophages. However, the work of Nicola Dalbeth and colleagues, reviewed in Chapter 5 , indicates that tophi are more dynamic structures than previously suspected, even in those in which typical acute inflammatory reactions do not occur. However, crystals are involved in degenerative reactions, which, over time, disrupt and alter the composition of extracellular matrix, producing irregular matrix heterogeneity resulting in decreased mechanical functionality. These reactions may be caused by crystal presence within cartilage or tendon insertion matrix or by chronic inflammation associated with crystals sequestrated in adjacent loose connective tissue spaces. This is described later in the sections on specific pathology of urate and CPPD crystals.
Gout (Monosodium Urate Crystal Deposition)
Since the discoveries of Garrod, it has been recognized that gout results from systemic disorders of uric acid metabolism, which manifest as hyperuricemia. However, it has also been recognized since the 19th century that patients may have hyperuricemia without gout, that acute gout can occur without hyperuricemia, that gout and osteoarthritis can coexist, and that urate crystals can be present on cartilage surfaces in the absence of episodes of acute gout. This indicates that local tissue urate supersaturation and other local factors must be present to enable urate crystal deposition. Crystals in acute gout and tophi have been definitively identified by x-ray diffraction analysis as monosodium urate monohydrate.
Acute Gouty Arthritis
In acute gout, MSU crystals appear in the synovial fluid, a local tissue space that becomes supersaturated in urate usually related to a systemic increase in urate in interstitial fluid and blood. MSU crystals seen in acute gout typically have a needle-shaped habit and are about 5 to 15 μm in length. In contrast to tophaceous gout, urate crystals in acute gout are distributed individually in random orientation.
The predilection of acute gouty arthritis for certain joints such as the big toe (podagra) and time of onset (night) illustrates the pathogenesis of acute urate crystal deposition. During the day, it is likely that small effusion develops in the first metatarsalphalangeal joint or adjacent bursa. At night, with elevation of the foot, the fluid drains. As urate diffuses through synovial tissues at less than 50% the rate of water, the urate concentration rises substantially. Another contributing factor may be decreased temperature, which lowers urate solubility. In the presence of supersaturation, minor trauma or a solid tissue component, such as collagen fragments, could serve to initiate crystal nucleation.
Acute gouty arthritis appears to be mediated by both the presence of crystals with elevated inflammatory potential in the synovial space, with additional triggers, such as elevated free fatty acids, helping to mediate induction of interleukin (IL)-1β secretion. Experimentally, the acute inflammatory reaction of model gout enters the cellular phase usually within a few hours of initial crystal injection. Single urate crystals are phagocytosed or partially phagocytosed principally by neutrophils. Later, smaller crystals are observed intracellularly and extracellularly. Unsuccessful phagocytic activity leads to inflammatory cell destruction with release of cell debris, lysosomal enzymes, and partially dissolved crystals, which further amplifies the inflammatory response and, later, crystal clearance. During the acute inflammation, the blood vessels in the synovium itself become dilated and congested. A neutrophil and fibrinous exudate can be observed within the synovium and in the SF. The SF can have cell counts well above 10,000 and can even simulate acute septic arthritis. While reported, coincident acute gout or pseudogout and septic arthritis is uncommon. Further, released lysosomal enzymes break down existing SF proteoglycan, decreasing urate crystal solubility and promoting additional crystal precipitation.
The acute inflammation is resolved by mechansms that include clearance of crystals via either dissolution or sequestration. As inflammation resolves, hyaluronate proteoglycan secretion from the synovium increases hydration and urate solubility. With crystal clearance, the synovial space and synovium are usually restored to its original state. With crystal sequestration that is present in recurrent acute gout, crystals tend to accumulate in the synovial recesses at the cartilage–synovium junction. Crystal persistence in this location can give rise later to tophaceous urate deposits and their cellular reaction, and be manifest by imaging.
Intercritical Gout
Intercritical gout refers to the quiescent period between attacks of acute gouty arthritis. It is now known that urate crystals can remain on the surface of articular cartilage during the intercritical period ( see Fig. 1-2 ). However, these crystals are present as aggregates. Release of these crystals into the synovial space is thought to produce only low-grade inflammation, because the crystals remain aggregated and coated with proteoglycan. Accordingly, one school of thought is that recurrent acute gouty arthritis with presence of individual urate crystals in random arrangement may reflect recurrent precipitation of urate in the synovial space. The other, and dominant, school of thought, reviewed in Chapter 5 , is that various triggers, including crystal “autoinjection” via crystal shedding (from synovial microscopic tophi and possibly cartilage surface deposits) and tophus remodeling by serum urate lowering and trauma, or proinflammatory triggers, are the primary stimuli of acute gouty inflammation. The latter model fits better with the fact that so many gout flares are triggered by drops rather than acute increases in serum urate. Moreover, many joint fluids in acute gout do not have large amounts of urate crystals on microscopic exam. As such, physical increases in the inflammatory potential of urate crystals in the synovium, and other inciting factors such as local or systemic inflammatory mediator generation, are held to be major drivers of acute gout attacks.
Chronic Tophaceous Gout
With the advent of effective urate-lowering drugs over the past 50 years, end-stage tophaceous gout, fortunately, is now often preventable. However, tophaceous gout remains a common presentation of the disease. Tophaceous gout can occur without previous history of acute gout, and urate crystal deposits with similar characteristics to tophi can be seen on articular cartilage surfaces of untreated patients with intercritical gout and even in some subjects with asymptomatic hyperuricemia. Excellent descriptions of tophaceous gout pathology and the effects of urate and urate crystal injections into articular tissues extend back in the scientific literature over 100 years.
In tophaceous gout, urate crystals precipitate in a matrix usually containing proteoglycans ( Figs. 1-3 to 1-7 ). Where crystals are uncovered, epitaxial precipitation of urate crystal aggregates is observed. These aggregates consist of needle-shaped crystals up to 20 μm × 1 μm × 1 μm aligned in parallel arrays (see Fig. 1-4 ). As the aggregates grow, groups of crystals become oriented to the characteristics of the tissue space. In the loose soft tissues such as bursa or synovium, the crystals may form spherical aggregates of crystal sheaves. On articular cartilage, the aggregates contain crystals aligned parallel to the joint surface. In both situations, urate crystals appear to bind extracellular proteoglycan as they are deposited.





The reactions to tophaceous urate deposits are very different depending on whether the crystals deposit in avascular or vascular tissue spaces. In avascular spaces such as articular cartilage surfaces, the crystal aggregates form first without any reaction of the underlying cartilage. With the accumulation of more urate crystals, necrosis of adjacent chondrocytes is seen (see Figs. 1-6 and 1-7 ). This pathophysiologic sequence is confirmed by old experimental evidence that urates do not deposit in necrotic tissues.
On morphologic observation, chondrocyte death subjacent to urate crystal deposits does not relate to mechanical pressure, osmotic effects of dissolved urate, or direct urate toxicity itself but likely results from interference with cell nutrition (nutrients must diffuse past the crystal aggregate barrier into the cartilage matrix). Regardless of the mode, chondrocyte necrosis results in failure to maintain the adjacent extracellular matrix. The matrix becomes physically weaker and has less capacity to retain water. At these sites, with time, these matrix features permit the crystal aggregates to enlarge and extend deeper into the cartilage, eventually reaching subchondral bone (see Fig. 1-7 ). At the most advanced stage, urate deposits can extend into the bone, replacing the trabecular spaces and eroding the cortical bone, eventually expanding to a nodular mass simulating tumor or osteomyelitis, and can advance across the joint space resulting in bony ankylosis. Rarely, fractures can occur through tophaceous bony deposits.
In contrast, where tophi form in connective tissue spaces that have a vascular supply, chronic inflammation characterized by C68 + macrophage, giant cell, and lymphocyte reaction is seen on the border (corona) of the tophaceous deposit (also discussed in Chapter 5 ). Without dissolution, chronic inflammation persists at the tophus–tissue interface (see Fig. 1-5 ). Successive generations of macrophages, unable to digest the crystal aggregates, die. Lipid from the residual cell membranes coalesces and precipitates to form cholesterol crystals, which in turn have surfaces that elicit macrophage and giant cell reactions as well as BCP calcification on the cholesterol crystal surfaces. Similar reactions to cholesterol crystals occur with atheroma. In advanced cases, clinical imaging can demonstrate the calcification surrounding the tophaceous deposits. On ultrasound, tophi are hyperechoic but may have a hypoechoic border, perhaps related to cholesterol crystal deposition.
Where urates become sequestrated in the synovial recesses at the joint margins, a synovial lining grows over the tophus, further isolating the crystals from the joint surface. However, the macrophage giant cell reaction persists and through cytokine production, particularly IL-1β, enables bone resorption and remodeling in the bone at the edge of the joint. This can result in joint erosions that superficially imitate those of rheumatoid arthritis (see Fig. 1-5 ). The erosions of tophaceous gout can be distinguished on imaging from those of rheumatoid arthritis by adjacent structural features. Unlike rheumatoid arthritis, there is no synovial hyperplasia or preferential erosion of articular cartilage at the joint margin; osteoporosis is not usually seen. With the treatment of gout, the erosions persist but contain loose fibrous connective tissue rather than crystals. The adjacent bone may demonstrate sclerosis.
Gouty tophi can form in any connective tissue but does have predeliction, perhaps because of lower temperature and decreased vascular circulation, for certain sites such as the helix of the external ear, the olecranon bursa, and extensor surface of the elbows. Clinical diagnostic confusion with rheumatoid arthritis is exacerbated by the frequent presence of rheumatoid factor in patients with tophaceous gout.
As noted earlier, gouty tophi can result in persistent chronic inflammation but can also result in an immune response. Immunoglobulin can adhere, through simple electrostatic interaction, to the highly negatively charged surface of urate crystals. Whether immune response to the immunoglobulin on the urate crystal surface, or other adjuvant effects of urate crystals on adaptive immunity, give rise to the presence of rheumatoid factor in patients with gout is not clear.
Some specific clinicopathologic presentations of tophaceous gout are noteworthy. Tophaceous gout can occur at acral sites, specifically the finger pads, with or without previous history of gouty arthritis. As well, tophaceous material can erode through skin, simulating pus from a sinus tract or an open wound. Not widely recognized, tophaceous gout can occur in the spinal intervertebral discs. Expansion of the tophus can lead to radiculopathy and cord compression.
Gout and osteoarthritis are common forms of arthritis and can coexist, but their pathologic features are entirely different. Nonetheless, the relative contribution of each disease to a patient’s arthritis can be a diagnostic challenge, particularly in patients with nodal osteoarthritis —tophaceous deposits can occur in or around Bouchard or Heberden nodes, as just one example.
Calcium Pyrophosphate Dihydrate Crystal Deposition Disease and Pseudogout
CPPD crystal deposition disease shares with gout the acute inflammatory reaction to the presence of crystals in the synovial space—hence, the clinical term pseudogout for the acute monoarticular arthritis presentation of the disease. The mechanisms of acute inflammation and resolution are also similar to those for urate crystals.
However, CPPD crystal deposition disease differs greatly from gout not only in the type of crystal formed but also in the tissue sites and mechanisms of crystal formation, the resulting clinical manifestations, and the associations with other diseases ( Fig. 1-8 ). This is reviewed in more detail in Chapter 20 , Chapter 21 .

CPPD crystal formation is restricted to only a few connective tissues: hyaline cartilage, fibrocartilage (meniscus, symphyses, entheses), intervertebral disc annulus, and, occasionally, fibrotic synovium with cartilaginous metaplasia ( Figs. 1-9 to 1-11 ). These tissues are avascular, with the cells receiving nutrition via diffusion. However, related to the high negative charge associated with the proteoglycan component, diffusion is limited to small molecules.



In blood, and in most connective tissues of the body, phosphatases, particularly alkaline phosphatases, are present. In these tissues, pyrophosphate (PP i ) secreted by cells or formed extracellullarly on the cell surface, rapidly hydrolyzes to an equilibrium concentration, PP i < 10 μmol/L. This concentration is far below the value of PP i > 200μmol/L necessary for CPPD crystal deposition.
Alkaline phosphatases formed on chondrocytes are bound to the outer cell membrane by phosphoinositol linkages. These high-molecular-weight molecules, when released from their phosphoinosotol linkages to the cell membrane by phospholipases, diffuse slowly, and only for limited distances, through the matrix in the affected tissues. Smaller molecules such as PP i can diffuse faster than the phosphatases, thereby escaping local hydrolysis by enzymes. PP i that has diffused beyond pyrophosphatase can become sequestrated in the matrix and increase to supersaturation concentration by coordinating to calcium bound to the matrix proteoglycans.
Crystal deposition is then initiated by local events such as increased impact force, delivering increased energy, local dehydration, local changes in concentrations of secondary ion (ions in the tissue environment not directly incorporated into the crystals), or local increase in PP i making the supersaturated state less stable.
As tissue Ca 2+ remains constant within narrow limits, local changes in extracellular secondary ions appear extremely important to CPPD crystal formation. Increased keratan sulfate–to–chondroitin sulfate ratio, commonly seen in aged cartilage, favors CPPD crystal formation, as does decreased Mg 2+ . Increased P i > 3 mmol/L strongly inhibits CPPD formation with the consequence that the formation conditions for CPPD and BCP crystals are mutually exclusive, a finding recently confirmed in matrix vesicle studies. Decreased P i as seen in hyperparathyroidism favors CPPD crystal formation. Secondary ions determine the kinetics of crystal formation and the exquisite crystal phase sensitivity of calcium pyrophosphate crystal formation. Of the more than 30 calcium pyrophosphate crystal phases, only two, calcium pyrophosphate dihydrate, monoclinic CPPD(M), and calcium pyrophosphate dihydrate, triclinic CPPD(T), form in biologic tissues. The increased association of CPPD crystal deposition disease with hypomagnesemia, hyperparathyroidism, and hypothyroidism may relate largely to secondary ion imbalances. The association of CPPD with hemochromatosis is more complicated and may relate to secondary hyperparathyroidism rather than direct effects of Fe 2+ ions.
As extracellular Ca 2+ is constant, CPPD crystal formation is dependent on increased extracellular pyrophosphate concentration ePP i . In turn, this relates to increased ePP i production by cell secretion or production on outer cell membrane as well as persistence of ePP i in the extracellular fluid. As reviewed in Chapter 20 , there are two sources for generation of extracellular PP i in the vicinity of the chondrocyte: PP i secretion from the chondrocyte via the phosphate pyrophosphate cell membrane transporter containing the ANKH protein and increased PP i generation at the cell membrane facilitated by the enzyme ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1). Both mechanisms are enhanced by upregulated ATP production.
Alkaline phosphatase is the key chondrocyte extracellular enzyme that promotes both PP i destruction and CPPD crystal dissolution, as it has PP i phosphatase activity at normal tissue pH. Alkaline phosphatase directly dissolves CPPD crystals by hydrolyzing PP i at the crystal surface ( Fig. 1-12 ). To have this effect, alkaline phosphatase must be in very close proximity or actually on the surface of CPPD crystals. Therefore, decreased alkaline phosphatase in the presence of increased ePP i are the prerequisite conditions for CPPD crystal formation in cartilage. At sites of CPPD crystal formation, alkaline phosphatase may be decreased because of insufficient enzyme synthesis or by enzyme inhibition from endogenous substances or by ePP i diffusion into the matrix beyond the reach of the enzyme. Similarly, without the presence of alkaline phosphatase in the immediate vicinity, CPPD crystal dissolution does not occur within cartilage matrix.

Selective inhibition of alkaline phosphatase activity by endogenous substances such as cysteine or mercaptopyruvate favors both increased extracellular PP i and inhibition of CPPD crystal dissolution. There is some early evidence that this mechanism can apply in vivo as tissue cysteine and mercaptopyruvate may be generated under hypoxic conditions as a response to free radical formation. As well, transforming growth factor (TGF)-β1 upregulation in chondrocytes by ionic ePi leads to increased ePP i and decreased alkaline phosphatase expression, two preconditions for CPPD crystal formation and inhibition of CPPD crystal dissolution Normal adult and aged mid-zone chondrocytes do not express alkaline phosphatase activity. Cartilage extracellular ionic Pi can become decreased in aging and various disease states, further setting up the local environment for CPPD crystal deposition ( Fig. 1-13 ).

In contrast to osteoarthritis, the chondrocytes in aged and CPPD crystal bearing cartilage do not appear to have as much hypertrophic differentiation or to be expressing significant alkaline phosphatase, which is a marker of chondrocyte hypertrophy. Some authors did consider CPPD-associated chondrocytes to be hypertrophic. However, this observation was relative to smaller superficial zone chondrocytes. Chondrocytes adjacent to crystals measured greater than 15 μm in diameter, which is within the upper normal range of human articular cartilage chondrocytes. Hence, chondrocyte hypertrophy linkage to CPPD crystal deposition is not fully established through differentiation analyses. Chondrocytes in the vicinity of CPPD crystals have been shown to be associated with increased lipid but the biologic significance in CPPD crystal deposition is as yet unknown.
Typically, in both hereditary and the more common sporadic disease, CPPD crystals first form extracellularly in the cartilage mid zone ( see Figs. 1-10 and 1-11 ). This is the tissue domain where the matrix is at the greatest distance from phosphatase activity in overlying SF or underlying bone. CPPD crystal deposition in both sporadic and hereditary disease is associated with matrix rich in keratan sulfate proteoglycan, a finding that is in accord with experimental studies. CPPD crystals precipitate as agglomerates of randomly oriented needle-shaped or rhomboidal crystals approximately 10 to 12 μm in longest dimension. While there is some experimental evidence suggesting that CPPD crystals may form in association with matrix vesicles (see Chapter 20 ), CPPD crystals in adult human articular cartilage form free in the extracellular space unassociated with cells, collagen, or matrix vesicles. Similar agglomerates can be formed in cell-free gels with high PP i concentrations simply through the differential diffusion of Ca 2+ ions. The smallest crystal agglomerate fills the space of a chondron, obliterating the chondrocyte. As the disease progresses, the agglomerates enlarge via epitaxial growth on existing crystals.
CPPD crystals are extremely insoluble in aqueous solution but can readily be dissolved by extracellular phosphatases including chondrocyte alkaline phosphatase. Polyamines, molecules found in cell nuclei, enhance CPPD crystal dissolution.
When the CPPD agglomerates reach sufficient size and proximity to the cartilage surface, the crystals are shed into the synovial space. Softening of cartilage matrix with increased hydration as may be seen in hypothyroidism will facilitate CPPD crystal shedding. CPPD crystals in the SF may be present without symptoms. However, CPPD crystals can induce low-grade cellular inflammation or incite an episode of acute inflammation, presenting clinically as acute pseudogout.
Similar CPPD crystal formation conditions are seen in meniscus, symphysis, or enthesis. However, CPPD crystals can also be observed in fibrocartilage overlying hyaline cartilage and in areas of chondrometaplasia in the meniscus, tendon insertions, flexor retinaculum and triangular cartilage of the wrist, annulus fibrosus, cartilage endplate and ligamentum flavum of the intervertebral disc, and occasionally synovium. Where the tissue environment is constrained by densely aligned collagen fibers such as tendon and fibrous synovium, CPPD crystals will tend to deposit in similar alignment. In more proteoglycan-rich environments such as cartilage, the crystals will precipitate in random agglomerates. Chondrometaplasia at these sites is associated with compressive forces and degeneration of tissues with fraying of collagen fibers and generation of matrix rich in proteoglycan.
CPPD crystal formation is to some extent species limited. CPPD crystals have been definitively identified in humans, monkeys, and dogs. Rabbits, rats, and mice form BCP crystals at the same sites. In part, these differences could be ascribed to the thickness of the crystal-bearing tissues, which have an increased distance from interstitial fluid phosphatase, and in part to differences in PP i metabolism between species.
Acute Pseudogout
In contrast to urate crystals that form in the SF, CPPD crystals are extruded into the synovial space from the adjacent hyaline articular cartilage or meniscal fibrocartilage. CPPD crystals incite acute inflammation via similar mechanisms to MSU crystals. In some cases, the synovial neutrophil count can exceed 50,000 cells/mm 3 and the arthritis can simulate septic arthritis. Phosphatases liberated from exudated neutrophils and phosphatase diffusing into the SF from permeable blood vessels likely act to dissolve CPPD crystals and help resolve the inflammation. As the inflammation progresses, the crystals become smaller and more difficult to recognize with light microscopy techniques.
Whatever the anatomic location, at each crystal deposition site, shedding of CPPD crystals into adjacent synovial or bursal space can incite acute inflammation. As well, CPPD crystals, like urate crystals, can be sequestered in synovial recesses and covered with fibrous tissue. Unlike urates, under most circumstances, the CPPD “pseudotophus” does not subsequently enlarge.
Chronic CPPD Crystal Deposition Disease
Unlike osteoarthritis (OA), which is a disorder with onset in middle age, CPPD crystal deposition disease commonly has onset after age 55. It also increases with aging, affecting more than 20% of the population older than 70 years, regardless of the joints affected, as reviewed in Chapter 21 . Pathologic CPPD crystal deposition disease is more common than that detected by radiologic chondrocalcinosis, since approximately 50% of the tissue density must be affected to observe chondrocalcinosis in a conventional radiograph. Chondrocalcinosis in radiologic OA is more closely associated with aging than with OA progression.
Considerable controversy exists about the overlap between the common diseases osteoarthritis and CPPD. CPPD crystals are observed occasionally in OA SF. In a longitudinal study, 25% of patients with knee joint OA developed CPPD. This approximates the prevalence of CPPD in elderly populations. CPPD crystal deposition in hyaline articular cartilage appears to be a distinct disease. Most surgically removed femoral heads that have been observed with CPPD crystal deposits have been removed for osteoporotic fracture. These femoral heads show intact cartilage thickness and articular surfaces rather than OA.
Two general patterns of CPPD crystal deposition are seen in knee joints removed for “degenerative” arthritis—a younger group with crystal deposition affecting predominantly meniscal fibrocartilage, coexistent with histologic OA, and an older group lacking cartilage radiologic and histologic features for OA but demonstrating CPPD arthropathy affecting fibrocartilage, hyaline cartilage, and synovium. With advanced arthritis, these patients typically present with CPPD crystals within the cartilage and extensive areas of sclerotic, eburnated bone articular surface. It remains to be determined whether or how much CPPD crystal deposits in cartilage stimulate cartilage matrix degradation via chondrocyte upregulation, “microcrystal-induced stress.” CPPD crystal deposits have very much different compliance from adjacent tissues and therefore can contribute to matrix degradation through physical wear.
Our observations are that some elderly patients with OA do have CPPD in knee meniscal fibrocartilage but only rarely is CPPD observed in hyaline cartilage affected by OA. As well, CPPD meniscal deposition is seen patients with OA associated with joint hypermobility. This distinction is important as typically CPPD diseased cartilage lacks the features of chondrocyte proliferation and increased chondrocyte alkaline phosphatase expression, characteristic changes seen in OA cartilage. As well, OA cartilage binds increased calcium, phosphorus, and magnesium compared to normal subjects, an ionic environment that inhibits CPPD calcification. From experimental evidence, episodes of pseudogout in OA patients may aggravate OA synovitis.
A second controversy relates to the simultaneous presence of CPPD and BCP crystals in OA SF. BCP crystals can be observed adjacent to CPPD crystal agglomerates, but this is very unusual as the formation conditions for these crystals are mutually exclusive. When this is seen, it implies that the crystals formed at different times. CPPD and BCP can be seen together in SF in advanced arthritis where a portion of the articular surface consists of exposed bone. Under these circumstances, BCP from the bone surfaces and CPPD from within cartilage can reach the SF.
While rheumatoid arthritis patients may have CPPD crystal deposition, CPPD also can present as pseudo–rheumatoid arthritis ( Chapter 21 ). As in chronic gout, patients may be seropositive for the rheumatoid factor. Pseudo–rheumatioid arthritis is characterized by extensive synovial deposition of CPPD crystals, accompanied by radiographic erosions similar to those in tophaceous gout. However, synovial inflammation is highly attenuated, and pannus formation is not observed.
CPPD crystal deposition is observed in the intervertebral discs and other soft tissue structures of the aged spine and can be a source of back or neck pain. CPPD crystal deposition at the cranial cervical junction such as in the transverse ligament of C1 and the periodontoid tissues can cause pain, myelopathy, and radiculopathy. Recently recognized radiologically, these patients can present with “crowned dens syndrome.” Of particular interest are linear and nodular CPPD deposits in the ligamentum flavum, which may account for the majority of cases of ligamentum flavum calcification commonly seen in Japan. As with CPPD deposition in other spinal tissues, large deposits with crystal shedding can lead to spinal cord compression and radiculopathy. The pathogenic relationship, if any, to the hierarchial culture of bowing remains to be understood. CPPD crystal deposits within tendons, like urate tophi, can lead to tendon rupture.
From clinical reports of CPPD following surgery for osteochondritis dissecans, meniscectomy, or intervertebral disc extrusion, CPPD crystals may form preferentially at sites of previous injury and fibrous repair.
CPPD has also been associated with rapidly destructive arthritis affecting the femoral head and other joints, predominantly in elderly woman. It is likely that this is a sequela of insufficiency fracture through the osteoporotic articular plate.
Familial CPPD crystal deposition disease is rare but has been much studied and is reviewed in Chapter 20 , Chapter 21 . From a pathology perspective, it is useful to classify familial CPPD into those families with CPPD and cartilage dysplasia, those with premature chondrocalcinosis younger than 30 years old, and those with familial occurrence as adults. Hypo-phosphatasia, where there is relative deficiency of alkaline phosphatase, is in the latter group and reinforces the role of decreased extracellular alkaline phosphatase activity in the pathogenesis of CPPD arthropathy.
CPPD arthropathy can present rarely as a “tumor” involving a single joint, raising the clinical and radiologic suspicion of neoplasia. As with the initially recognized case, more than 50% of known cases involve the temporomandibular joint. The pathogenesis of this condition is unknown but typically the connective tissue cells in the vicinity of the crystal deposits exhibit chondroid metaplasia.
Related posts:
 Diagnosis of Gout
Synovial Fluid Crystal Analysis
Uricosuric Therapy of Hyperuricemia in Gout
Diet, Alcohol, Obesity, Hyperuricemia, and Risk of Gout
Pathogenesis and Molecular Genetics of Calcium Pyrophosphate Dihydrate Crystal Deposition Disease
Plain Radiography and Advanced Imaging of Calcium Pyrophosphate Dihydrate Crystal Deposition Disease and of Arthropathy Associated With Basic Calcium Phosphate Crystal Deposition
Diagnosis of Gout
Synovial Fluid Crystal Analysis
Uricosuric Therapy of Hyperuricemia in Gout
Diet, Alcohol, Obesity, Hyperuricemia, and Risk of Gout
Pathogenesis and Molecular Genetics of Calcium Pyrophosphate Dihydrate Crystal Deposition Disease
Plain Radiography and Advanced Imaging of Calcium Pyrophosphate Dihydrate Crystal Deposition Disease and of Arthropathy Associated With Basic Calcium Phosphate Crystal Deposition
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


