Polyglutamine repeat expansion in the androgen receptor is responsible for the motor neuron degeneration in X-linked spinal and bulbar muscular atrophy (SBMA; Kennedy’s disease). This mutation, like the other polyglutamine repeat expansions, has proven to be toxic itself by a gain-of-function effect; however, a growing body of evidence indicates that loss of androgen receptor normal function simultaneously contributes to SBMA disease pathology, and, conversely, that normal androgen receptor signaling mediates important trophic effects upon motor neurons. This review considers the trophic requirements of motor neurons, focusing upon the role of known neurotrophic factors in motor neuron disease natural history, and the interactions of androgen receptor signaling pathways with motor neuron disease pathogenesis and progression. A thorough understanding of androgen receptor signaling in motor neurons should provide important inroads toward the development of effective treatments for a variety of devastating motor neuron diseases.
The androgen receptor (AR) is a nuclear hormone receptor of approximately 110 kd that is responsible for the biological actions of androgens, including testosterone and its metabolite dihydrotestosterone (DHT). Androgens regulate a wide range of developmental and physiologic processes including the growth of muscle and bone, spermatogenesis, and the development of secondary sexual characteristics . The AR mediates these effects primarily by influencing the expression of androgen responsive genes. The AR affects transcription in a ligand-dependent manner by nuclear translocation, dimerization, DNA binding, and association with various co-activators/co-repressors and other components of the transcriptional machinery . The functional domains of the AR – an N-terminal transcriptional activation domain, a DNA-binding domain (DBD) and a C-terminal ligand-binding domain ( Fig. 1 ) – reflect these processes involved in androgen signaling. Both gain-of-function and loss-of-function mutations have been identified in the AR, and these alterations respectively give rise to distinct pathologies. These conditions include the various forms of androgen insensitivity syndromes (AIS): ranging from testicular feminization (Tfm) and partial androgen insensitivity syndrome (PAIS) to male infertility, prostate cancer (CaP), breast cancer susceptibility, and X-linked spinal and bulbar muscular atrophy (SBMA), which is also known as Kennedy’s disease . SBMA is of particular interest because it exhibits characteristics of both loss-of-function and gain-of-function mutations in the AR.
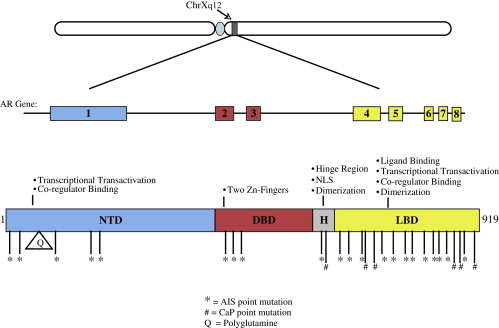
SBMA is a heritable, adult onset disease that causes preferential degeneration of lower motor neurons leading to weakness and atrophy of bulbar, facial, and limb muscles . It is clinically similar to amyotrophic lateral sclerosis (ALS), another form of motor neuron disease (MND). The main clinical distinction between the two diseases is that ALS involves degeneration of both upper and lower motor neurons, whereas the affected cell type in SBMA is lower motor neurons. Another interesting difference is that Onuf’s nucleus, an androgen-sensitive spinal cord motor neuron nucleus, is spared in ALS, although it degenerates in SBMA . Degeneration of sensory neurons of the dorsal root ganglia is also a typical sign associated with SBMA, often preceding the onset of motor dysfunction . In addition to the neurologic phenotype, SBMA patients display some of the characteristic signs of androgen insensitivity syndromes including testicular atrophy, decreased fertility, gynecomastia, and elevated androgen levels . This mild androgen insensitivity, along with the X-linked pattern of inheritance, caused the AR to be considered as a candidate gene for SBMA. This association was further supported by linkage mapping of the SBMA gene to a locus on the X chromosome that includes the AR gene . Ultimately, the work to identify the gene responsible for SBMA led to the discovery of a novel mutation, the expansion of a trinucleotide CAG repeat, in the first exon of the AR gene . This CAG repeat encodes a stretch of glutamine residues within the N-terminal transcriptional activation domain of the AR. Unaffected individuals have a polyglutamine repeat size that ranges between 5 and 35 glutamines, while symptomatic individuals always have a polyglutamine stretch of at least 37 glutamines . This absolute association indicates that the polyglutamine expansion in the AR is the source of the pathology in SBMA. SBMA is a member of a family of nine dominantly inherited neurodegenerative diseases caused by polyglutamine (polyQ) repeat expansions in specific proteins. Other polyQ diseases include Hungtington’s disease (HD), dentatorubral-pallidoluysian atrophy, and six forms of spinocerebellar ataxia. Interestingly, though each of the polyQ diseases has a CAG repeat expansion in a different gene, and each disease seems to specifically affect different subsets of neuronal populations, SBMA is the only polyQ repeat expansion disease known to cause selective degeneration of motor neurons . This observation implies that there is something unique about AR biology that is important for motor neuron function.
Studies of other polyQ expansion diseases, as well as research specifically investigating SBMA, indicate that the pathology associated with polyQ-expanded proteins is due to a toxic gain of function of the mutant protein . The polyQ expansion of the AR in SBMA, like all polyQ repeat expansion diseases, is a dominantly inherited mutation; numerous lines of investigation, including ascertainment of homozygous females with CAG repeat expansions and androgen ablation experiments in SBMA transgenic mouse models , have shown that SBMA disease pathogenesis requires AR activation by ligand binding. Heterozygous female carriers are thus spared the neurodegenerative SBMA phenotype due to a significantly reduced concentration of available ligands for the AR. Although considerable work in the polyQ research field has emphasized the polyQ protein misfolding gain-of-function toxicity as the principal mechanism for molecular pathology, gain-of-function and loss-of-function are not mutually exclusive explanations for the etiology of the disease. Indeed, numerous studies have implicated loss of polyQ protein normal function in the pathogenesis of each disease . In the case of SBMA, one of the primary indicators of a loss of normal AR function is the mild androgen insensitivity phenotype that is seen in affected patients. This insensitivity reflects the fact that polyQ-expanded AR is not competent to fully stimulate androgen-dependent physiologic pathways or to fully maintain male secondary sexual characteristics. Consequently, in a motor neuron-like cell culture model of SBMA, cells transfected with polyQ-expanded AR differentially express genes in response to androgens when compared with cells expressing a non-pathologic form of AR , confirming that polyQ-expanded AR is not functionally interchangeable with unexpanded AR. In a highly representative SBMA transgenic mouse model, the authors’ lab has further demonstrated that endogenous mouse AR partially compensates for, and thus retards, motor neuron loss, as SBMA mice develop a more severe motor neuron phenotype when the endogenous AR is absent . These observations indicate that normal AR might be playing a role in supporting motor neurons and protecting them from degeneration. Hence, polyQ-expanded AR is dysfunctional in this protective capacity, an effect that agonizes the process of motor neuron degeneration in SBMA. This article examines the evidence supporting the idea that normal AR has a protective, trophic role in motor neuron biology, first by examining the trophic requirements of motor neurons in general, and then by delineating the trophic properties of the AR in the central nervous system.
Trophic requirements of motor neurons
The neuronal requirements for trophic factor support are well established and have been investigated for over 50 years. Nerve growth factor (NGF) was the first target-derived neuronal growth factor identified. Since its discovery in 1951, a whole family of similar trophic factors, called neurotrophins, have been recognized . Neurotrophins are small peptide growth hormones that promote neuronal differentiation, synaptic plasticity, regeneration, and survival . The family includes molecules such as neurotrophin-3 (NT-3), brain-derived neurotrophic factor (BDNF), ciliary neurotrophic factor (CNTF), and glial cell line-derived neurotrophic factor (GDNF) . Neurotrophin signals are transduced in a variety of ways, but most involve the activation of receptor tyrosine kinases. NGF, BDNF, and NT-3 involve signaling through the Trk receptors (ie, TrkA, TrkB, and TrkC) and also through the p75 NTR (neurotrophin receptor). GDNF signals through the RET family of receptor tyrosine kinases and CNTF signals through a receptor that is structurally very similar to the IL-6 receptor.
Of all of these neurotrophic factors, GDNF, BDNF, and CNTF display the greatest growth promotion and neuroprotective effects on motor neuron populations. GDNF is produced by glial cells of the CNS; is a very potent neuroprotective agent; and exerts its effects on both astrocytes and motor neurons. Mutations in murine GDNF are associated with a loss of 20%–30% of motor neurons , whereas GDNF over-expression prevents the developmental programmed cell death of motor neurons and axotomy-induced motor neuron cell death . Muscle from patients who have SBMA and ALS demonstrates decreased expression of GDNF . BDNF is target-derived and retrogradely transported by motor neurons from muscle . BDNF can also protect motor neurons from axotomy-induced cell death , as well as prevent toxic neuronal nitric oxide synthase production and glutamate excitotoxicity . Despite these protective effects, BDNF has shown little promise in the treatment of several MNDs, including ALS , though it did ameliorate the disease phenotype in the Wobbler mouse . CNTF is a peptide growth factor that is produced predominantly by glial cells postnatally, but, unlike the other neurotrophins, does not contain a signal sequence, indicating that it is not normally secreted . This has led some to hypothesize that CNTF release occurs in response to nerve injury . Despite some conflicting reports , CNTF appears to rescue axotomized neurons from cell death . In addition, both homozygous null CNTF mice and mice deficient in the CNTFα receptor develop motor neuron loss.
Beyond these classical neurotrophic factors, there are several other growth factors that display motor neuron specific trophism, including vascular endothelial growth factor (VEGF) and insulin-like growth factor 1 (IGF-1). VEGF is a cytokine that is typically associated with angiogenesis and can support motor neurons by increasing local blood supply, but also exhibits many direct neuroprotective effects on motor neurons . VEGF is able to promote the survival of motor neurons in vitro , and increase neurogenesis after axotomy . Decreased VEGF levels have been correlated with various forms of MND including ALS, both in patients and in mouse models, including the authors’ transgenic mouse model of SBMA . Intriguingly, deletion of a hypoxia-responsive element within the promoter of VEGF leads to the degeneration of motor neurons and a correlation between VEGF promoter haplotypes and ALS has been observed . In addition, VEGF treatment has been beneficial in a variety of models of MND .
IGF-1 is a peptide hormone produced by oligodendrocytes, Schwann cells, and muscle . IGF-1 is a very potent trophic factor for motor neurons; it increases motor neuron survival both in vitro and in an in vivo axotomy model . IGF-1 also promotes motor neuron axonal sprouting, regeneration, and muscle innervation in vitro . Targeting of IGF-1 to motor neurons slows the typical force decline seen in aging muscle . Because IGF-1 is trophic for both motor neurons and muscle, there has been considerable interest in its potential protective capacity in various forms of MND. Most notably, viral delivery of IGF-1 dramatically slowed the progression of ALS in the SOD1 transgenic mouse model . Additionally, the beneficial effects of IGF-1 in the Wobbler mouse model of MND are potentiated by glycosaminoglycans , and the neuroprotective effects of IGF-1 and GDNF act additively in the SOD1 mouse model of ALS . IGF-1 can decrease the toxicity associated with polyQ-expanded AR in vitro by stimulating the Akt phosphoylation of the AR, thereby preventing ligand binding and receptor activation . These trophic factors play an important role in protecting motor neurons from many of the cytotoxic insults that lead to motor neuron cell death.
Trophic effects of the AR
The trophic effects of androgens on reproductive organs are well established. AR signaling is essential for the normal development of male sexual features, including sexual organ development as well as the formation and maintenance of secondary sexual characteristics, including increased body hair and muscle mass . Beyond sexual development, androgen signaling has been associated with neoplastic prostate growth and male breast carcinoma , which demonstrates the strong trophism of androgens. Hormonal castration, the prevention of androgen signaling, and silencing prostate AR expression have all been demonstrated to retard cancerous growth of the prostate . In this context, the trophic effects of androgens have been causally linked with the ability of the AR to stimulate VEGF expression, thereby increasing angiogenesis. However, androgens, as well as other sex hormones, have also demonstrated trophic actions in non-reproductive tissues. Immunohistochemical investigation has confirmed the presence of the AR in a wide variety of human fetal extra-genital tissues including the thymus, bronchial epithelium, cardiac valves and surrounding muscle, and in the spinal cord . The presence of the AR in these early tissues suggests that androgen signaling might not simply affect sexual differentiation, but may be a broader signal for early growth and development of these tissues.
It is clear that AR expression in the spinal cord occurs as early as the first trimester and that high expression of AR within the spinal cord continues into adulthood. In a classic paper in 1977, Sar and Stumpf demonstrated that androgen receptor expression is especially high in the ventral spinal cord. Therefore, it is reasonable to predict that AR signaling is important to the development and maintenance of this tissue, including especially spinal cord motor neurons. Studies of cell culture and animal models of spinal cord development have provided clues to the functions of androgens in such cell types. AR-expressing motor neuron-like cells in vitro exhibit changes in morphology in response to androgen treatment, including developing larger cell bodies and broader fields of neurite processes . Further exploration of these effects indicates that this increased neurite outgrowth might be due to androgen-dependent up-regulation of neuritin, a protein previously demonstrated to be important for neurite elongation . Another indication of the function of androgens in developing spinal cord motor neurons comes from careful investigation of the spinal nucleus of the bulbocavernosus (SNB), a sexually dimorphic nucleus of androgen-sensitive motor neurons in rodents that is analogous to Onuf’s nucleus in humans. Androgen signaling has been demonstrated to influence soma size and dendrite length in SNB motor neurons . These effects are linked to the expression of both BDNF and its receptor TrkB . Furthermore, androgens and CNTF interact early in postnatal development to increase SNB motor neuron number . These in vivo data confirm the in vitro observation that androgen signaling plays a crucial role in motor neuron development, especially with regard to the establishment of neurites (in vitro) and dendrites (in vivo). However, remaining questions are: to what degree does the AR maintain this trophic influence in adult motor neurons, and how might androgen signaling protect adult motor neurons from degeneration?
Trophism of adult neurons is frequently inferred from the ability of a trophic factor to protect neurons from a variety of insults. The AR has demonstrated a wide variety of neuroprotective effects. AR signaling protects many cells (including neuroblastoma cells , primary cerebellar granule cells and striatal cells in vivo ) against oxidative stress-induced cell death. This protection may result from an AR-specific increase in catalase activity . Another example of AR neuroprotection comes from an experiment in which the excitotoxin kainate was noted to kill more hippocampal neurons in gonadectomized male rats versus sham-operated controls; this cell death was then reversed by supplementation of the gonadectomized rats with DHT . Androgens have even been shown to protect neurons from the toxic effects of the beta-amyloid peptide (Aβ1-42), believed to be a causative agent in Alzheimer’s disease pathogenesis . Both androgens and estrogen can protect neurons from Aβ toxicity by increasing the levels of the chaperone protein Hsp70, which is thought to prevent Aβ aggregate formation . Additionally, androgens may protect against Aβ toxicity through a receptor-dependent activation of the MAPK/ERK signaling pathway in neurons, leading to downstream activation of Rsk and inactivation of Bad, a pro-apoptotic Bcl-2 family member .
Importantly, in the spinal cord, signaling through the AR can elicit specific motor neuron protection, both in vitro and in vivo. AR expressing motor neuron-like MN-1 cells not only exhibit larger somas and broader neurite arbors in response to androgens, but they are also protected from serum deprivation-induced cell death . This seems to be a common function of AR signaling in neurons, as androgens promote the survival of human primary neurons in low serum conditions in a receptor-dependent fashion . Evidence from in vivo motor neuron injury models indicates that AR signaling protects motor neurons from axotomy-induced death. Androgens interact with BDNF in the SNB to promote motor neuron survival and maintenance of the SNB dendritic arbor post-axotomy . Furthermore, androgens are particularly trophic in a hamster facial motor neuron axotomy model, promoting survival of axotomized neurons and increasing the rate of functional recovery . AR signaling is clearly involved in protecting adult neurons, including motor neurons, from toxicity, injury and death.
How might androgen signaling be promoting neuronal survival? Since the AR functions primarily as a transcription factor, it is logical to predict that this protective effect might be tied to the ability of the AR to induce the expression of genes that are involved in promoting motor neuron survival ( Fig. 2 ). Indeed, AR mediated up-regulation of neuritin expression is directly responsible for neurite elongation in motor neuron-like cultures, as neuritin silencing by siRNA abolishes the trophic response to androgens . Similarly, inhibition of neurotrophin and CNTF signaling prevents the sparing of SNB motor neurons normally observed after administration of androgens to neonatal rats . Trophic factor signaling is thus an important aspect of AR trophism and might function downstream of androgen signaling.
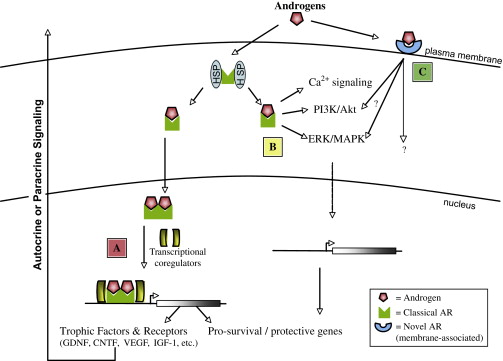
Several trophic factors that are important for motor neuron biology have been associated with AR signaling. AR activation stimulates the production of VEGF mRNA in prostate cancer cells ; and in vivo siRNA silencing of AR has been demonstrated to slow neoplastic prostate growth and repress VEGF expression . AR stimulation of VEGF signaling increases BDNF levels and ultimately neurogenesis in the brains of adult songbirds , which demonstrates the possible multi-potency of AR-stimulated VEGF expression. Transgenic mice carrying a polyQ-expanded AR express less VEGF, and this reduction has been implicated in the pathology of SBMA . In another mouse model of SBMA, polyQ-expanded AR is associated with decreased GDNF and IGF-1 expression . Researchers have identified androgen response elements in the upstream promoter of the IGF-1 gene , and testosterone has been observed to stimulate IGF-1 transcription in prostate . Interestingly, IGF-1 can facilitate AR signaling by de-repressing Foxo1 or by stabilization of beta-catenin, an AR co-activator . The authors’ group, in collaboration with workers at the National Institutes of Health, has recently identified a mechanism through which IGF-1 prevents the deleterious effects of polyQ-expanded AR by stimulating Akt-mediated AR phosphorylation which blocks ligand binding and receptor activation . These results indicate that while IGF-1 and AR signaling in motor neurons are intertwined, it is possible that AR stimulation of IGF-1 expression is a protective mechanism for motor neurons. CNTF is another important trophic factor that has been demonstrated to be androgen-responsive. Expression of the CNTF receptor alpha subunit is regulated by the AR in both spinal cord and muscle . Furthermore, CNTF receptor knock-out mice do not exhibit the sexual dimorphism normally present in the SNB ; and SNB motor neuron death in androgen-insensitive rats can be prevented by CNTF administration . These results, together with the previously noted observation that blocking CNTF signaling prevents the AR-mediated rescue of early SNB motor neurons, indicate that AR-mediated expression of the CNTF receptor is necessary for the sexually dimorphic sparing of SNB motor neurons in rodents. These data support a model in which AR-mediated expression of trophic factor signaling genes is a mechanism by which androgens convey their neuroprotection.
A complication of this interpretation comes from recent observations that AR signaling might not be restricted to direct genomic interaction and the subsequent regulation of transcription through binding of androgen response elements in the promoters of target genes (See Fig. 2 ). Indeed, it has been reported that the androgen antagonists flutamide and cyproterone acetate both exhibit agonist effects, including hippocampal neuroprotection, in an AR-dependent manner . These antagonists have been shown to block AR DNA binding and disrupt AR co-regulator associations, respectively, ultimately preventing the transcriptional effects of the AR . This suggests that either these compounds are not true anti-androgens or, more likely, that signaling through the AR is not limited to influencing transcription. Indeed, the AR, like many hormone receptors , can rapidly signal through ERK/MAPK and the PI3K/Akt pathways without direct genomic interaction . These interactions could promote cell survival, however, rapid signaling of the AR through these second messenger pathways has not been directly observed in motor neurons. In addition, Gatson and colleagues recently described the discovery of a novel membrane-associated androgen receptor action in a glial cell model. This membrane-associated receptor, triggered by a cell-impermeable BSA-conjugated DHT, was shown to have the opposite effect upon MAPK and Akt signaling pathways that had been elicited by non-BSA-conjugated DHT treatment. This group then further characterized their system in primary astrocyte cultures, and demonstrated that signaling through this potentially novel androgen pathway actually promotes astrocyte cell death . These data indicate that despite years of research, there may be numerous aspects of AR signaling that are not fully understood.
Trophic effects of the AR
The trophic effects of androgens on reproductive organs are well established. AR signaling is essential for the normal development of male sexual features, including sexual organ development as well as the formation and maintenance of secondary sexual characteristics, including increased body hair and muscle mass . Beyond sexual development, androgen signaling has been associated with neoplastic prostate growth and male breast carcinoma , which demonstrates the strong trophism of androgens. Hormonal castration, the prevention of androgen signaling, and silencing prostate AR expression have all been demonstrated to retard cancerous growth of the prostate . In this context, the trophic effects of androgens have been causally linked with the ability of the AR to stimulate VEGF expression, thereby increasing angiogenesis. However, androgens, as well as other sex hormones, have also demonstrated trophic actions in non-reproductive tissues. Immunohistochemical investigation has confirmed the presence of the AR in a wide variety of human fetal extra-genital tissues including the thymus, bronchial epithelium, cardiac valves and surrounding muscle, and in the spinal cord . The presence of the AR in these early tissues suggests that androgen signaling might not simply affect sexual differentiation, but may be a broader signal for early growth and development of these tissues.
It is clear that AR expression in the spinal cord occurs as early as the first trimester and that high expression of AR within the spinal cord continues into adulthood. In a classic paper in 1977, Sar and Stumpf demonstrated that androgen receptor expression is especially high in the ventral spinal cord. Therefore, it is reasonable to predict that AR signaling is important to the development and maintenance of this tissue, including especially spinal cord motor neurons. Studies of cell culture and animal models of spinal cord development have provided clues to the functions of androgens in such cell types. AR-expressing motor neuron-like cells in vitro exhibit changes in morphology in response to androgen treatment, including developing larger cell bodies and broader fields of neurite processes . Further exploration of these effects indicates that this increased neurite outgrowth might be due to androgen-dependent up-regulation of neuritin, a protein previously demonstrated to be important for neurite elongation . Another indication of the function of androgens in developing spinal cord motor neurons comes from careful investigation of the spinal nucleus of the bulbocavernosus (SNB), a sexually dimorphic nucleus of androgen-sensitive motor neurons in rodents that is analogous to Onuf’s nucleus in humans. Androgen signaling has been demonstrated to influence soma size and dendrite length in SNB motor neurons . These effects are linked to the expression of both BDNF and its receptor TrkB . Furthermore, androgens and CNTF interact early in postnatal development to increase SNB motor neuron number . These in vivo data confirm the in vitro observation that androgen signaling plays a crucial role in motor neuron development, especially with regard to the establishment of neurites (in vitro) and dendrites (in vivo). However, remaining questions are: to what degree does the AR maintain this trophic influence in adult motor neurons, and how might androgen signaling protect adult motor neurons from degeneration?
Trophism of adult neurons is frequently inferred from the ability of a trophic factor to protect neurons from a variety of insults. The AR has demonstrated a wide variety of neuroprotective effects. AR signaling protects many cells (including neuroblastoma cells , primary cerebellar granule cells and striatal cells in vivo ) against oxidative stress-induced cell death. This protection may result from an AR-specific increase in catalase activity . Another example of AR neuroprotection comes from an experiment in which the excitotoxin kainate was noted to kill more hippocampal neurons in gonadectomized male rats versus sham-operated controls; this cell death was then reversed by supplementation of the gonadectomized rats with DHT . Androgens have even been shown to protect neurons from the toxic effects of the beta-amyloid peptide (Aβ1-42), believed to be a causative agent in Alzheimer’s disease pathogenesis . Both androgens and estrogen can protect neurons from Aβ toxicity by increasing the levels of the chaperone protein Hsp70, which is thought to prevent Aβ aggregate formation . Additionally, androgens may protect against Aβ toxicity through a receptor-dependent activation of the MAPK/ERK signaling pathway in neurons, leading to downstream activation of Rsk and inactivation of Bad, a pro-apoptotic Bcl-2 family member .
Importantly, in the spinal cord, signaling through the AR can elicit specific motor neuron protection, both in vitro and in vivo. AR expressing motor neuron-like MN-1 cells not only exhibit larger somas and broader neurite arbors in response to androgens, but they are also protected from serum deprivation-induced cell death . This seems to be a common function of AR signaling in neurons, as androgens promote the survival of human primary neurons in low serum conditions in a receptor-dependent fashion . Evidence from in vivo motor neuron injury models indicates that AR signaling protects motor neurons from axotomy-induced death. Androgens interact with BDNF in the SNB to promote motor neuron survival and maintenance of the SNB dendritic arbor post-axotomy . Furthermore, androgens are particularly trophic in a hamster facial motor neuron axotomy model, promoting survival of axotomized neurons and increasing the rate of functional recovery . AR signaling is clearly involved in protecting adult neurons, including motor neurons, from toxicity, injury and death.
How might androgen signaling be promoting neuronal survival? Since the AR functions primarily as a transcription factor, it is logical to predict that this protective effect might be tied to the ability of the AR to induce the expression of genes that are involved in promoting motor neuron survival ( Fig. 2 ). Indeed, AR mediated up-regulation of neuritin expression is directly responsible for neurite elongation in motor neuron-like cultures, as neuritin silencing by siRNA abolishes the trophic response to androgens . Similarly, inhibition of neurotrophin and CNTF signaling prevents the sparing of SNB motor neurons normally observed after administration of androgens to neonatal rats . Trophic factor signaling is thus an important aspect of AR trophism and might function downstream of androgen signaling.
Related posts:
 Glial Cells in ALS: The Missing Link?
Glial Cells in ALS: The Missing Link?
 Motor Unit Number Estimation in the Assessment of Performance and Function in Motor Neuron Disease
The Role of Exercise in Amyotrophic Lateral Sclerosis
Motor Unit Number Estimation in the Assessment of Performance and Function in Motor Neuron Disease
The Role of Exercise in Amyotrophic Lateral Sclerosis
 Designing Clinical Trials in Amyotrophic Lateral Sclerosis
Cognitive and Behavioral Impairment in Amyotrophic Lateral Sclerosis
Clinical Trials in Spinal Muscular Atrophy
Designing Clinical Trials in Amyotrophic Lateral Sclerosis
Cognitive and Behavioral Impairment in Amyotrophic Lateral Sclerosis
Clinical Trials in Spinal Muscular Atrophy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


