The long-established study of osteogenesis imperfecta (OI) has opened a realm of scientific research surrounding connective tissue disorders. Over the past decade alone there have been vast advancements in the understanding of the underlying genetic variations of this disease, pharmacologic treatments, and the technological and surgical options for fracture deformity. It is important to appreciate the progressive nature of the advances concerning OI. This article aims to synthesize the expanding evolution of the field surrounding OI over the past decade.
Key points
- •
Osteogenesis imperfecta (OI) can be a debilitating disease with a wide range of phenotypic manifestations.
- •
The Sillence subtypes of OI can be explained by different variations in genetic mutations.
- •
Bisphosphonate therapy augments bone turnover and increases bone density in OI patients, although its efficacy in preventing fracture, reducing pain, and improving function is controversial.
- •
Recommendations for indications, duration, and type of bisphosphonate therapy in children have not been agreed upon.
- •
Fassier-Duval rodding systems show great promise in decreasing fracture rates while requiring less revision and causing fewer complications traditionally seen with older systems.
The science and genetics of osteogenesis imperfecta
History
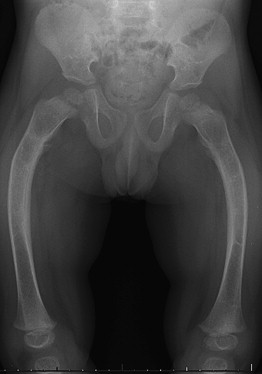
The first scientific documentation of osteogenesis imperfecta (OI) by army surgeon Olaus Jacob Elkman dates back to 1788. He detailed observational reports of bone fragility and fractures in 3 familial generations afflicted with “congenital osteomalacia.” OI was later given its name by Dutch professor Willem Vrolik in 1849. The varying array of clinical manifestations grouped under this broad diagnosis sparked a field of study that aimed to classify and determine the root cause of this debilitating disease ( Fig. 1 ). David Sillence developed his classification system in the 1970s, that has since been added to and modified, but is still widely used today ( Table 1 ).
| OI Type | Gene Defect | Inheritance |
|---|---|---|
| I | COLA1 | AD |
| II | COLA1/COLA2 | AD |
| III | COLA1/COLA2 | AD |
| IV | COLA1/COLA2 | AD |
| V | Not known | AR |
| VI | SERPINF1 | AR |
| VII | CRTAP | AR |
| VIII | LEPRE1 | AR |
| IX | PPIB | AR |
| X | SERPINH1 | AR |
| XI | FKB910 | AR |
| N/A | PLOD2 | AR |
| N/A | LRP5 | AR |
| N/A | SP7 | AR |
Past Discovery
Electron microscopy research in OI patients in the 1970s led to the discovery of altered collagen structure in these patients compared with that of normal histologic controls. This collagen was branded Type I, as it was the first discovery of altered connective tissue leading to clinical disease. In the 1980s, a gene deletion coding for the pro-α1 collagen chain steered the discovery of poor collagen synthesis and inspired further research detailing the underlying genetic causes of OI. As a result, it was thought for a long time that mutations in collagen type I genes (COLA1 or COLA2) were the sole cause of OI, and that it was an autosomal dominant disorder.
Current Genetics
In the early 1990s, Wallis and colleagues described cases of OI not caused by mutations in COLA1 or COLA2. Stemming from this study, there has been the discovery of multiple new forms of OI not originally described by Sillence. These forms involve various other genetic defects that manifest in an autosomal-recessive fashion. These genetic defects involve CRTAP, LEPRE1, PPIB, SERPINH1, SERPINF1, PLOD2, FKBP10, LRP5, and SP7 (see Table 1 ).
The science and genetics of osteogenesis imperfecta
History
The first scientific documentation of osteogenesis imperfecta (OI) by army surgeon Olaus Jacob Elkman dates back to 1788. He detailed observational reports of bone fragility and fractures in 3 familial generations afflicted with “congenital osteomalacia.” OI was later given its name by Dutch professor Willem Vrolik in 1849. The varying array of clinical manifestations grouped under this broad diagnosis sparked a field of study that aimed to classify and determine the root cause of this debilitating disease ( Fig. 1 ). David Sillence developed his classification system in the 1970s, that has since been added to and modified, but is still widely used today ( Table 1 ).
| OI Type | Gene Defect | Inheritance |
|---|---|---|
| I | COLA1 | AD |
| II | COLA1/COLA2 | AD |
| III | COLA1/COLA2 | AD |
| IV | COLA1/COLA2 | AD |
| V | Not known | AR |
| VI | SERPINF1 | AR |
| VII | CRTAP | AR |
| VIII | LEPRE1 | AR |
| IX | PPIB | AR |
| X | SERPINH1 | AR |
| XI | FKB910 | AR |
| N/A | PLOD2 | AR |
| N/A | LRP5 | AR |
| N/A | SP7 | AR |
Past Discovery
Electron microscopy research in OI patients in the 1970s led to the discovery of altered collagen structure in these patients compared with that of normal histologic controls. This collagen was branded Type I, as it was the first discovery of altered connective tissue leading to clinical disease. In the 1980s, a gene deletion coding for the pro-α1 collagen chain steered the discovery of poor collagen synthesis and inspired further research detailing the underlying genetic causes of OI. As a result, it was thought for a long time that mutations in collagen type I genes (COLA1 or COLA2) were the sole cause of OI, and that it was an autosomal dominant disorder.
Current Genetics
In the early 1990s, Wallis and colleagues described cases of OI not caused by mutations in COLA1 or COLA2. Stemming from this study, there has been the discovery of multiple new forms of OI not originally described by Sillence. These forms involve various other genetic defects that manifest in an autosomal-recessive fashion. These genetic defects involve CRTAP, LEPRE1, PPIB, SERPINH1, SERPINF1, PLOD2, FKBP10, LRP5, and SP7 (see Table 1 ).
Diagnosis of OI
Prenatal
Our understanding of this disease from a genetic level has not only allowed for more accurate diagnosis of the disease using collagen molecular testing, but also has allowed us to understand the manifestations of the disease, even as early as the prenatal period. Features that can be seen on prenatal ultrasonography usually between 14 and 18 weeks of gestation (for Types II and III OI; Type I cannot be easily diagnosed) include increased nuchal translucency, reduced echogenicity of bones, multiple fractures of the long bones, ribs, and skull at various stages of healing, and bowing of the long bones (+/− shortening). Once there is a concern for OI via prenatal ultrasonography, the diagnosis can be made via either (1) chorionic villus sampling demonstrating abnormal type I collagen via electrophoresis, or (2) amniocentesis, which obtains fetal DNA for molecular analysis. As the majority of mutations will involve COLA1/COLA2 genes, testing is centered around the identification of these genes followed by examination of the other aforementioned genes.
Postnatal
In the postnatal period, positive clinical findings (multiple fractures, blue sclera, and so forth) and the exclusion of other metabolic causes of osteoporosis can then warrant confirmation of the diagnosis via dermal biopsy and/or DNA analysis. Dermal biopsy has been shown to be about 90% positive in suspected OI cases, whereas DNA analysis has been able to identify COLA1/COLA2 mutations in 95% of cases of OI, with the remaining percentage having CRTAP/LEPRE1 mutations. As a result, it may be suggested that DNA testing should be utilized because it is more sensitive for disease diagnosis. Best-practice guidelines exist for the laboratory diagnosis of OI.
Treatment of OI
An understanding of the genetics of OI allows for an insight into the current molecular methods of disease diagnosis, which have evolved tremendously since the clinical descriptors of pathology used initially. Once the diagnosis has been made, the treating team (including the orthopedic surgeon) must formulate the appropriate treatment plan for the patient. These interventions include both pharmacologic and surgical interventions.
Pharmacologic Treatment
Basic science
The primary objective of OI therapy revolves around decreasing the number of pathologic fractures, decreasing pain, increasing growth, improving bone metabolism, and optimizing function. The current mainstay of pharmacologic treatment has been centered on bisphosphonate (BP) therapy, which serves to decrease bone turnover and inhibit bone resorption. Within the adult population receiving treatment for osteoporosis, it is clear that BPs slow down bone resorption by shortening osteoclast life. This fact creates a dilemma in the child treated with OI: although less bone is resorbed, osteoblasts may still be producing defective collagen. In turn, this leads to several quandaries regarding the treatment of pediatric OI patients with BPs: the potential long-term side effects in children, whether changes in bone mineral density (BMD) can lead to improved bone strength and/or bone matrix, the most effective dosing/duration/mode of administration for the various forms of OI and varying ages, and whether fracture risk and function are improved with treatment. Although current guidelines do not exist, there have been several key studies in the literature detailing the results of BP therapy in children with OI.
Clinical systematic reviews
In a Cochrane review, Phillipi and colleagues examined the results of BP treatment of OI. The investigators looked specifically at BMD, fracture reduction, and improvement in clinical function, reflective of the knowledge surrounding BP and OI treatment. Only 8 quality studies were identified. From these studies, it was concluded that both oral and intravenous bisphosphonate therapy increased BMD in children and adults with OI. However, this increase in BMD was not shown to translate into decreased fracture risk or improvement in clinical parameters (pain, growth, and/or mobility).
These findings were also supported by the work of Castillo and colleagues in their own systematic review. Whereas improvement in BMD was found with BP treatment, decreased fracture rate and improved growth once again could not be found. In addition, the reviewed studies were lacking in their examination of the effect of treatment on deformity, need for surgery, pain, function, and quality of life. Further recommendations regarding dosing regime and treatment duration were also unclear, as was the impact of therapy on patients with mild OI or infants.
Even with the aforementioned limitations, several studies have attempted to answer these questions, and these need to be examined specifically to aid in understanding the best pharmacologic treatment options, with further high-quality study still being necessary in the future.
Oral BP treatment
Oral treatment was initially the mode of BP delivery for patients with bone disorders. Sakkers and colleagues examined 34 pediatric patients with OI who were randomly assigned to placebo or oral olpadronate for 2 years. The investigators found that olpadronate treatment was associated with a 31% reduction in the relative risk of long-bone fracture and an increase in spinal bone mineral content and density. However, there was no difference in functional outcome, anthropometrics, vertebral height, or urinary markers of bone resorption.
Furthermore, Ward and colleagues examined alendronate for the treatment of OI in a randomized, placebo-controlled study. In this multicenter study, 130 children with Types I, III, or IV OI were randomized to placebo or alendronate for 2 years. Alendronate was found to increase bone mineral density by 51% as opposed to 12% by placebo ( P <.001), and bone turnover (measured by urinary N-telopeptide of collagen I) decreased by 62% in the alendronate group versus 32% in the placebo group ( P <.001). However, the incidence of long-bone fracture, bone pain, and physical activity were similar between the alendronate and placebo groups, similar to the findings of the systematic reviews already cited.
Intravenous BP treatment
Intravenous treatment developed as an alternative to oral BP, with potential increased potency and speed of action. Barros and colleagues compared the safety and efficacy of zoledronic acid and pamidronate in children with OI. Pamidronate has been proposed as the standard treatment in children with OI. In 23 patients with OI over a 1-year period, intravenous infusion was used for these medications. Both groups had statistically significant increases in BMD (67.6% and 51.8% respectively) with minimal side effects.
Intravenous BP treatment versus oral BP therapy
In one of the few studies comparing intravenous BPs with oral BPs, DiMeglio and Peacock compared oral alendronate and intravenous pamidronate in children with OI in a 2-year randomized controlled trial. Eighteen children participated in the study, which found that total body and lumbar spine BMD increased, turnover markers decreased, and linear growth increased equally in both groups. Fracture incidence was not significantly different between both groups. These studies still leave the question unanswered as to the best method of treatment for OI patients, and the potential short-term and long-term consequences of the particular mode of treatment chosen by the clinician.
Role of growth hormone with BP therapy
Some investigators have postulated that the use of BP with recombinant human growth hormone (GH) may be a feasible treatment for some forms of OI, particularly as GH may increase the production of type I collagen in patients who have a collagen synthesis defect. Antoniazzi and colleagues performed a randomized controlled trial comparing children who underwent neridronate treatment alone or neridronate with GH. Patients who underwent combined treatment had greater improvement in BMD and growth velocity, although there was no difference in fracture risk.
BP treatment, orthopedic intervention, and quality of life
One of the main concerns regarding BP treatment is that although it has been shown to reliably increase BMD, the translation of this increased BMD into improved quality of life and a decreased need for orthopedic intervention has not been definitively established. Looking at a more clinically relevant set of outcomes, de Graaff and colleagues examined outpatient visits and operative interventions in patients who underwent BP therapy. The investigators retrospectively examined OI patients undergoing BP therapy from 1988 to 2009 to determine the clinical efficacy and outcomes of pharmacologic intervention. Of the 201 OI patients in this study, 118 were treated with BP along with vitamin D and calcium supplementation. The primary end points were the number of outpatient visits by each patient; this rate decreased from 3.35 to 2.14 per year. There was also a decrease in operative fracture interventions, from 0.73 per year to 0.38 per year. Regarding the overall group, clinic visits naturally decreased with age (also in the nontreatment group). However, after the age of 7, visits decreased significantly with BP therapy.
Quality of life was also examined by Kok and colleagues, whose results differed from those of de Graaff and colleagues. In this study, 34 children with OI were randomized to olpadronate or placebo. Quality of life was measured using the Self-Perception Profile for Children and the Health-Utility Index. The investigators found only slightly significant improvements in quality of life from BP treatment, with no significant differences with regard to pain.
These studies once again raise the concern of BP treatment being able to translate microstructural changes (increased BMD) into macro–clinically relevant outcomes.
Treatment of infants and mild OI patients with BP
Although BP treatment is used in older severely affected patients, it is unclear whether patients at the extremes, infants who are severely affected with OI, or older patients with mild OI can benefit from BP treatment.
Regarding the treatment of young children, Antoniazzi and colleagues prospectively examined the efficacy of BP treatment in infants with severe OI (Type III). Five children who started treatment with intravenous neridronate at birth, 5 children who started intravenous treatment at 6 months of age, and a historical control group of 10 children with OI who were untreated were compared. The investigators found that patients who were started with intravenous treatment after birth had a lower incidence of fractures than the other 2 groups in the first 6 months of treatment. In addition, the birth-treated group had the highest improvement in vertebral body area and structure.
The long-term deleterious effects of BP treatment are unknown; therefore, BP treatment of “mild” OI is still controversial. Rauch and colleagues examined the use of BPs in patients with mild OI. Twenty-six children with mild OI (type I) were randomized to oral risedronate or placebo for 2 years. The investigators found that the risedronate group had decreased levels of bone-resorption markers (35% vs 6%), and increased lumbar spine BMD Z-scores (increase of 0.65 vs a decrease of 0.15). However, there was no difference in the number of new fractures or bone mass and density of the total body.
Further study is necessary to determine the role of treatment with BPs in the various forms of OI, with consideration of their potential clinical utility and long-term side effects in children.
Surgical Management
Along with the ever-expanding knowledge of the genetic basis of OI and pharmacologic treatments, there have also been major advancements in surgical technique and technology addressing fracture treatment and intervention in OI. Among the constellation of symptoms and maladies that plague patients with OI, the principal issue from an orthopedic standpoint remains pathologic fractures and deformities of long bones (see Fig. 1 ). With poor bone stock and decreased quality of collagenous tissues, patients can experience fractures starting in utero and later in life from otherwise benign falls or minor injuries. There has been a continual evolution of the fixation options for these patients.
Early treatment modalities
In 1959, Sofield and Millar introduced the concept of osteotomies secured with intramedullary nails to treat fractures and deformity in patients with OI. Treatment involved complete subperiosteal exposure of the bone with multiple osteotomies fixed around an intramedullary pin. Although leading to excellent correction of deformity and fracture prevention, this procedure was fraught with complications including devascularization of bone via a wide subperiosteal exposure, bone thinning, decreased ambulatory capacity, hardware failure, and multiple revisions because of growth.
As a result, this led to the development of expanding rod systems, most notably developed by Bailey and Dubow ( Fig. 2 ). These rods had fixation in the epiphyses (via a T-piece) and grew as the child grew. These early telescoping systems, however, had high complication and failure rates. Lang-Stevenson and Sharrard examined the results of 28 patients who underwent fixation with Bailey-Dubow extensible rods and found 10 instances of proximal migration of the distal end of the rod, 1 incorrect placement in the proximal femur, 4 instances of loosening of a T-piece, and 3 infections in the area of the rods. Similar complications were noted by Jerosch and colleagues in their review of 107 long-bone fixations with Bailey-Dubow rods. Here there was a 63.5% complication rate, most commonly rod migration combined with perforation of the joint, bone, and soft tissues.






