Fig. 1.1
Microarray SNP genotyping and copy number analysis. a Individual SNP genotypes are determined by the fluorescent color emitted when allele-specific probes (shown in black) are hybridized to the sample DNA (shown in blue). Each allele is assigned a color (red or green). Homozygous genotypes fluoresce the color of the respective allele, while heterozygous genotypes fluoresce yellow. Millions of SNPs can be genotyped simultaneously on a single microarray. b Copy number is determined from the intensity of the fluorescent probes. Signal intensity is plotted as the logR ratio, which is the log-scale of the sample’s intensity divided by the expected signal intensity. Higher logR denotes copy number gains or amplifications, while drops in logR signal deletions (shown). c Copy number variations can be summarized and plotted for all chromosomes with the chromosome ideogram.
Copy Number Variation
CNV microarrays contain millions of probes that use fluorescent detection methods to determine the patient’s genotype at millions of SNPs throughout the genome (Fig. 1.1a). Briefly, DNA is hybridized to the microarray and the genotype of each SNP is detected using fluorescent probes. The signal intensity correlates with the amount of DNA present in the sample. Therefore, regions of the genome with lower than expected signal intensities are designated as deletions and regions with higher than expected signal intensities are designated as amplifications. Individual probe data can be visualized using log-ratio plots. Each SNP probe marker is plotted along the chromosome to show the log-scaled ratio (logR) of the patient’s probe intensity compared to the expected intensity (Fig. 1.1b). For a normal copy number (N = 2), the logR is equal to 0. The logR ratio increases and decreases in the presence of amplifications and deletions, respectively. Multiple computational programs are available to identify CNVs from the individual probe-level data, and CNVs may be plotted alongside the human ideogram (Fig. 1.1c) .
CNVs Associated with Orthopaedic Disorders
Charcot-Marie-Tooth (CMT) 1 is a class of hereditary demyelinating neuropathies characterized by decreases in nerve conduction with secondary foot and ankle difficulties caused by muscle weakness, hip dysplasia and spinal deformities, including scoliosis, kyphoscoliosis and thoracic kyphosis [9]. CMT type 1A, often seen in orthopaedic clinics, was one of the first diseases associated with a submicroscopic duplication [10, 11]. Specifically, a 1.5 Megabase (Mb) duplication of chromosome 17p11.2 harboring the PMP22 gene is the primary cause of CMT1 and was reported in majority of CMT1 patients [12]. It is worth noting that the discovery of the CMT1A duplication inspired a new term, “genomic disorder”, to describe genetic diseases arising not from simple sequence changes but from complex rearrangements of the genome [13]. Today, molecular genetic clinical testing for the 17p11.2 duplication is widely available and provides a definitive diagnosis of CMT1A.
Recently, a genome-wide CNV study in individuals with idiopathic clubfoot identified a microduplication of the TBX4 gene in multiple cases [14], and together with copy number variation of the PITX1 gene [15], these CNVs implicate the intricate processes of hindlimb development in idiopathic clubfoot [16]. The developmental processes leading to proper bone formation, including proper morphology of supporting structures (muscles, tendons), involve complex regulatory mechanisms with precise spatio-temporal expression of a multitude of genes. The effect of CNVs on gene expression has been well documented [17]. Therefore, it is plausible that CNVs play a substantial role in conditions commonly ascertained in orthopaedic clinics.
Homozygosity Mapping
SNP genotypes from microarray experiments can also be analyzed to identify regions of homozygosity (ROH), that is, parts of the genome that are devoid of typical genetic variation (Fig. 1.2a). ROH regions result when both copies of the chromosome are identical and every SNP in the region is homozygous. Rarely does the human genome contain long stretches of homozygosity; ROH up to ~ 3 Mb in length occur naturally in the general population [18]. The primary cause of extended ROH (> 3 Mb) in a patient is consanguinity; generational inbreeding decreases the amount of total variation in a person’s genome and increases the amount of homozygosity in the genome (Fig. 1.2b). Because consanguineous parents share a common ancestor, regions of their genome are the same. Therefore, those same regions will be identical in their children. All SNPs in those regions will be homozygous; thus, as the parents are more closely related (or as generational inbreeding continues), ROH regions expand further across the genome. The lack of genetic variation within a consanguineous family or population leads to a higher occurrence of rare recessive disease. Therefore, disease-gene mapping in consanguineous families focuses on identifying large ROH regions, and, probabilistically, disease-causing homozygous mutations frequently occur in the largest ROH region. Homozygosity mapping is a powerful method for identifying disease genes in consanguineous families/populations and is easily detected using commercial SNP microarrays.

Fig. 1.2
Regions of homozygosity in consanguineous families. a Microarray SNP genotypes are plotted along the chromosome using the B-allele frequency. The B-allele frequency is the measured intensity of the “B” allele divided by the total signal intensity of the SNP (either homozygous AA & BB or heterozygous AB). Homozygous SNP B-allele frequencies are near 0 and 1, and heterozygous SNPs are near 0.5. When plotted along the chromosome, large regions of homozygosity can be easily identified (shown by green bar). b Regions of homozygosity occur frequently in consanguineous families and result in a higher occurrence of rare recessive disorders. Children from a first-cousin mating may present with increased incidence of a disorder (filled symbols) than would be expected by chance.
Homozygosity Mapping in Orthopaedic Disorders
The gene causing autosomal recessive Horizontal Gaze Palsy with Progressive Scoliosis (HGPPS) was identified using homozygosity mapping in consanguineous families. Homozygosity mapping in two consanguineous families initially narrowed the disease locus to chromosome 11q23–25, as it was the only region homozygous in all affected individuals in both families and not homozygous in the unaffected family members [19]. Subsequently, ten HGPPS consanguineous families with homozygosity at the chromosome 11q23–25 region were studied to refine the candidate gene region and identify the disease-causing gene [20]. Sequence analysis identified rare homozygous mutations in the ROBO3 gene in all families. All homozygous mutations identified in affected family members were not present in control subjects; therefore, the likelihood of these mutations being homozygous in the general population is very low. This study of HGPPS families highlights the power of homozygosity mapping in consanguineous families with rare recessive disease.
In another example, ROH mapping in consanguineous families was used to identify homozygous mutations causing CMT type 4C (CMT4C) [21]. This study identified a region on chromosome 5 that was homozygous in affected individuals from two consanguineous Turkish families. A candidate region was identified as homozygous only in affected individuals and heterozygous in the unaffected siblings, and mutation analysis identified seven different homozygous mutations in the SH3TC2 gene in nine consanguineous families with CMT4C.
Recessive diseases require mutations in both copies (alleles) of the gene. Thus, mutations are inherited from both unaffected parents, and because consanguineous parents share a common ancestor, both parents share the same disease-causing mutation. Recessive diseases are generally caused by sequence variants that render the encoded protein non-functional, so called “loss-of-function” variants. Loss-of-function changes may be frameshift, nonsense, or splice-site mutations that change the amino acid sequence of the protein (frameshift), prematurely truncate the protein (nonsense) or cause entire regions of the protein to be removed (splice-site) (Fig. 1.3). Notably, because of their highly deleterious affects, these mutations occur less frequently in the genome compared to other types of mutations (e.g. synonymous mutations that do not change the protein sequence or missense mutations that change only a single amino acid), have a lower population frequency (are rare) and are more frequently associated with disease [22]. In the CMT4C study for example, five of the seven (71 %) mutations were either frameshift mutations or splice-site mutations [21]. Other mutations were missense mutations that changed single amino acids in the protein, but the consequences of these mutations on protein function were not immediately obvious.
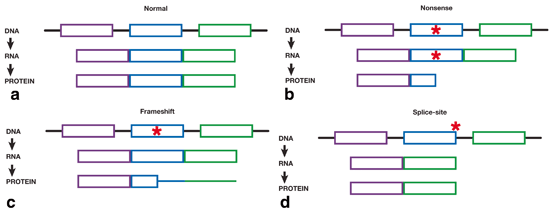
Fig. 1.3
Schematic diagram showing the effect of mutations on RNA splicing and changes to the resultant protein. a DNA consists of exons (colored sections) and introns (black bars). Exons are transcribed into RNA and the introns are excluded. Finally, each exon is translated into the final protein, with each exon corresponding to a specific region of the protein. The full protein is represented by all exons being transcribed and translated fully (represented by boxes). b Nonsense mutations (red star) result in a truncated protein. All exons are fully transcribed; however, translation is prematurely halted and the protein is shortened. All protein sequence after the mutation is lost. c Frameshift mutations (red star) affect the translation of the protein. All protein sequence prior to the mutation is normal (shown as boxes); however, the sequence after the frameshift mutation is different and results in a protein with different sequence and length (shown by colored bars). d Splice mutations (red star) occur at the beginning or end of introns and results in entire exons being skipped during transcription. When an exon is skipped during transcription, the same exon is not translated into the protein. Thus, the entire region of the protein is lost.
Next-Generation Sequencing
Next-generation sequencing , or NGS, has revolutionized genomics research by providing a lower-cost and faster alternative to traditional sequencing methods, enabling high-throughput whole-genome and whole-exome sequencing. Because of this new and efficient technology, the 1000 Genomes (1000G) Project set out to catalogue normal sequence variation with at least a 1 % frequency in many world-wide populations [23]. The resulting 1000G database (http://browser.1000genomes.org/index.html) is an important resource for the genetics community, as it provides the ability to distinguish common benign variation from potentially disease-causing variants. Thus, coupling NGS and public resources like 1000G provides researchers with new opportunities for discovering genes associated with human disease. Beyond research applications, clinical opportunities for NGS are under investigation, as described later in this chapter [24].
Whole-Exome Sequencing
The exome is defined as the protein-coding regions of the genome. The exome corresponds to exons, the parts of genes that encode for RNA and/or protein, and represents about 1–2 % of the entire genome. Whole-exome sequencing (WES) utilizes NGS technology and is an attractive alternative to whole-genome sequencing (WGS); therefore, it is a widely used method to identify genetic causes of rare disease [25]. The goal of WES is to identify disease-causing variants that change the protein sequence (also called nonsynonymous variants) or that alter the function of a RNA molecule. The first WES study to identify new disease-causing variants identified nonsynonymous mutations in the MYH3 gene in four individuals with dominant Freeman-Sheldon syndrome (FSS), also called distal arthrogryposis type 2A [26]. As detailed below, this study established proof of concept for the power of WES to discover disease-causing variants with relatively few study subjects. This rare-variant analysis strategy has since been used repeatedly to identify pathogenic mutations for many rare human disorders [25, 27]. For a detailed discussion of WES analysis and application to orthopaedic conditions, the readers are directed to a recently published review [28].
Rare Variant Exome Analysis
In the era preceding WES, disease gene discovery depended on access to extended families with multiple affected individuals, suitable for inheritance mapping. However, because WES examines virtually every gene simultaneously, it has the potential to identify disease-causing genes without the need for large family studies. In this way, WES may identify a single disease-associated gene by studying multiple unrelated individuals with the same rare phenotype. This type of analysis seeks to identify a single, or at least only a small number, of candidate genes (of the ~ 20,000 in the genome) with rare or novel mutations shared among all affected individuals. Rare mutations are typically defined as those mutations that occur in < 1 % of the general population, as determined from the 1000G database. Novel mutations are those that are not identified in the 1000G database (or other databases) nor in other control individuals. As a group, rare and novel nonsynonymous mutations occur at evolutionarily conserved amino acids in the protein and, therefore, may be deleterious and alter the protein’s function. Thus, they are more frequently associated with human disease [22, 29]. As well, a rare-variant analysis assumes that the same gene is causing the disease in all affected individuals, although the mutation does not need to be identical. Therefore, because the same gene is expected to harbor a rare or novel variant in all affected individuals, the number of candidate genes will decrease as more cases are sequenced. In the proof of concept study by Bamshad et al., four unrelated cases with dominant FSS as well as 8 unrelated control samples were sequenced [25]. Combining WES results from all four cases identified a single gene, MYH3, with novel deleterious disease-causing mutations that were absent in controls [26]. In a separate study, rare-variant analysis was applied to WES of four cases with recessive Miller syndrome from three families (two of the cases were siblings). Candidate genes were identified using a recessive model that required two novel mutations. Ultimately, this study identified a single gene, DHODH, with novel recessive mutations in all affected individuals [30].
These early successes suggested that WES in small cohorts might be sufficient to identify rare or novel nonsynonymous disease-causing mutations. This is critically important for studies of rare orthopaedic disorders, where large numbers of unrelated affected cases are difficult to ascertain. The number of unrelated cases needed to identify a small number of candidate genes is determined, in part, by the frequency of the disease. If the disease is very rare, it is unlikely to be caused by common mutations; therefore, it may be appropriate to apply a WES analysis that considers only novel mutations. If the disease is modestly rare or uncommon, WES analysis may consider a frequency cutoff that only considers novel mutations and mutations with a < 1 % population frequency. The relationship between the number of resulting candidate genes, analysis strategy and number of cases is shown in Fig. 1.4. For very rare disorders in which only novel variants are considered, sequencing more than one affected individual has a dramatic effect on the number of candidate genes (333 versus 33), and sequencing even more cases further decreases the number of candidate genes, although to a lesser degree (33 versus 5).
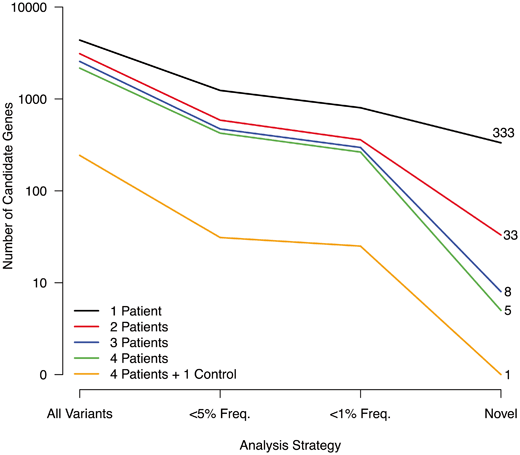
Fig. 1.4
Number of candidate genes by analysis and number of patients. The number of candidate genes from whole-exome sequencing are shown for different numbers of patients. As the number of patients increases, the number of mutations shared by all patients decreases (Y-axis). As well, the number of candidate genes decreases as the analysis strategy includes only rare or novel mutations (X-axis). Thus, studies of rare disorders in which multiple patients are sequenced may identify a small number of candidate genes.
The power to identify a small number of candidate genes is greater for recessive compared to dominant disorders because a recessive disorder requires mutations in both copies of the gene. Multiple independent mutations in the same gene occurs much less frequently in the population, and accordingly WES of fewer cases will provide sufficient power to detect fewer candidate genes. This concept was demonstrated using WGS in a single patient with recessive disease [31]. Of > 600 genes with novel nonsynonymous mutations, only 42 (~ 7 %) genes had multiple mutations, and only one of the 42 genes had two loss-of-function (nonsense) mutations. This study presumably would have identified the same mutations with WES and demonstrates the power of these technologies to identify recessive disease-causing genes from a small number (or even a single) of affected individuals.
Family-Based Exome Analysis
Analysis of WES in families is an alternative strategy to sequencing unrelated cases. For families with multiple affected relatives, this analysis method provides multiple advantages compared to the rare-variant approach. First, the possibility of genetic heterogeneity (disease caused by different genes in different individuals) is removed. In the presence of high genetic heterogeneity, the rare-variant analysis strategy has limited power to identify the disease-causing gene(s) in unrelated individuals, as was recently demonstrated in a WES study of Kabuki syndrome [32]. In this study, no single gene harbored novel loss-of-function mutations in all ten cases [32]. However, the gene MLL2 contained novel loss-of-function mutations in nine of the ten cases. Ultimately, the study identified MLL2 mutations in 66 % of Kabuki cases. Using a family-based approach, all affected relatives are expected to share the same disease mutation, therefore, WES in families provides greater power to overcome genetic heterogeneity.
Related posts:
 The Genetic Architecture of Idiopathic Scoliosis
The Genetic Architecture of Idiopathic Scoliosis
 Molecular Genetics of Congenital Multiple Large Joint Dislocation
Molecular Genetics of Congenital Multiple Large Joint Dislocation
 Neurofibromin in Skeletal Development
Neurofibromin in Skeletal Development
 DMP-1 in Postnatal Bone Development
DMP-1 in Postnatal Bone Development
 Genetic and Environmental Interaction in Malformation of the Vertebral Column
Genetic and Environmental Interaction in Malformation of the Vertebral Column
 Somatic Mutations in Overgrowth Syndromes
Somatic Mutations in Overgrowth Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


